What is Genome Mapping? Genome mapping is defined as assigning a specific location to a specific gene on a particular
What is Genome Mapping and What Alignment Tools are There?
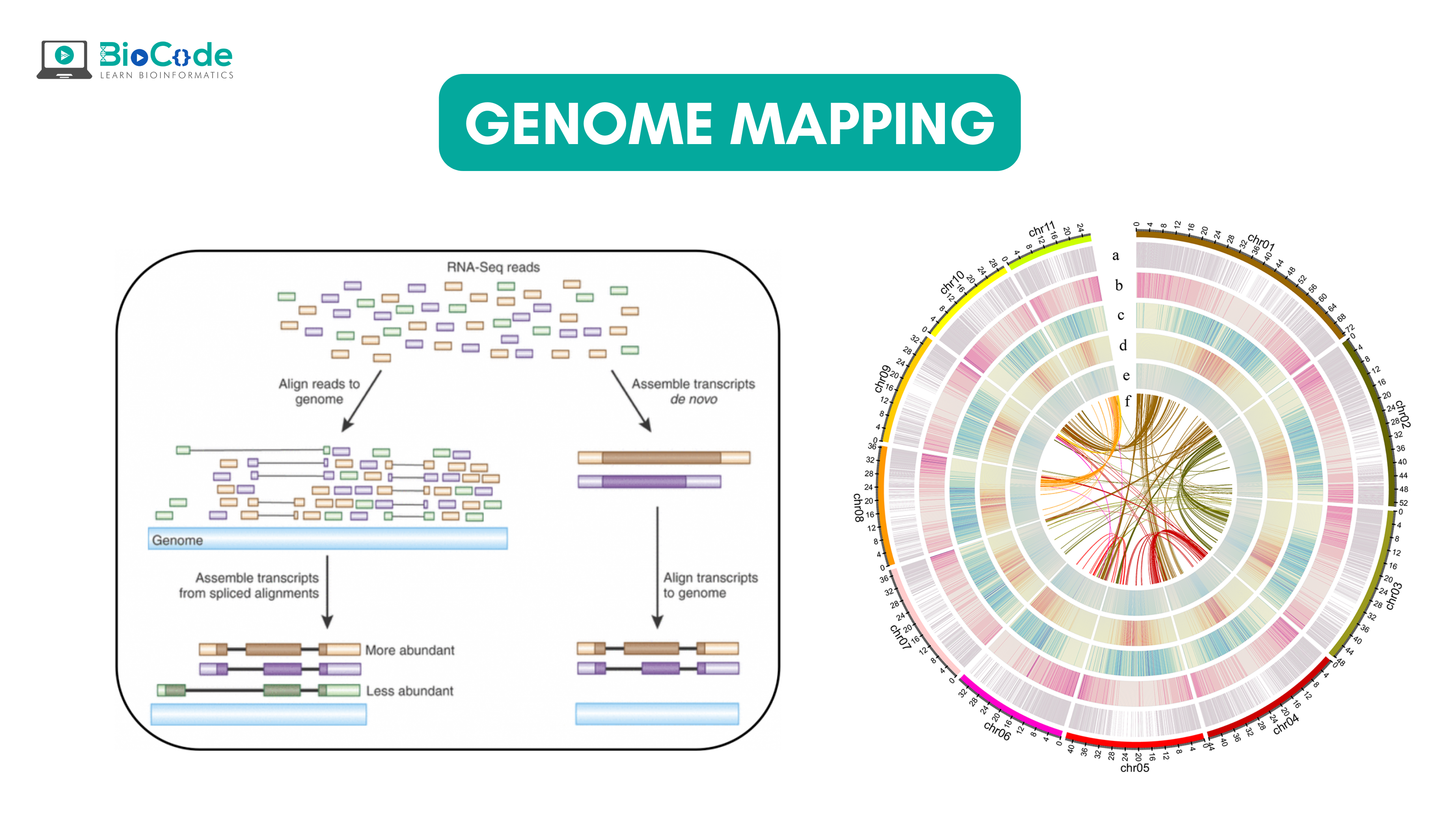
What is Genome Mapping? Genome mapping is defined as assigning a specific location to a specific gene on a particular
