
Introduction
New pharmaceutical drugs are developed through a systematic process of “Drug Discovery”. We know that the recent decade has seen the development of medical sciences-related computational tools and algorithms, some of them are also extended to drug discovery. The implementation of computations techniques in drug discovery is known as “Computer-aided drug design (CADD)”. For instance, cheminformatics is the use of computer science to understand and characterize molecular and chemical attributes of some specific compounds. Many of the bioinformatics tools are now being used to characterize many small molecules and generate libraries so that they can be screened for specific therapeutic processes.
In addition to this, bioinformatics techniques have been used to understand how these small molecules are causing therapeutic activity in human beings, this includes the interaction of drugs with proteins, the impact of drugs on biological pathways, and illuminating those genetic variations that can modify the drug response.
Position of CADD in the drug discovery pipeline
In a normal drug discovery pathway, the potential candidate molecule gives a “hit” during the screening process based upon its binding with the target protein. Through CADD, the rate of “hit” is enhanced because it carries out more targeted research than the classical drug discovery pathway. It also aims to identify the potential derivative that can improve the activity of the drug. In a drug discovery pipeline, CADD is used in three main stages or steps:
Filtering of libraries having a large number of compounds into a predicted small set of active compounds which can be verified experimentally.
Optimization of lead compounds to increase affinity or improve drug metabolism and pharmacokinetics (DMPK) properties like absorption, distribution, metabolism, excretion, and the potential for toxicity (ADMET)
Designing of a novel compound
The CADD can be categorized into two main categories:
Structure-Based CADD
Ligand-Based CADD
But before going into detail about these two main types, it is important to discuss the ligand database.
Ligand Databases for CADD
For virtual high-throughput screening in order to screen a large number of chemical compounds as a potential “hit” for our drug, it is necessary to have a virtual library or a database. Currently, different hardware and softwares allow the screening of approximately 100,000 molecules each day through parallel processing clusters, this can be done only in the presence of virtual libraries.
The virtual screening libraries are of a variety of sizes. They can be general libraries that are used against any target for screening and they are developed by using several different computational tools and algorithms. Earlier, the structure generation constraint was only the molecular formula i.e., the number of atoms in a molecule of a compound. But as the comprehensive computational tools were developed, it became difficult to enumerate all possible conformations, thus further restrictions were applied. Also, the chemical compounds whose synthesis was impossible or were known or predicted to cause negative effects on DMPAK/ADMET were excluded. In addition to this, Fink and his colleagues proposed a method for the construction of virtual libraries in which connected graphs were used which were populated with N, C, O, and F atoms, and then they were trimmed based on structural constraints and elimination of unstable structure. The database which was proposed by using this method was named “Generated a DataBase (GDB)” and it has almost 26.4 million chemical structures of compounds that are used for vHTS (virtual high-throughput screening). Later, this database was a bit modified and known as GDB-13 which includes C, N. S, O, and Cl and has almost 970 million compounds.
Some of the popularly used repositories of chemical compounds along with their contents are given in Table 1:
Table 1: Chemical Compounds Libraries and their Content
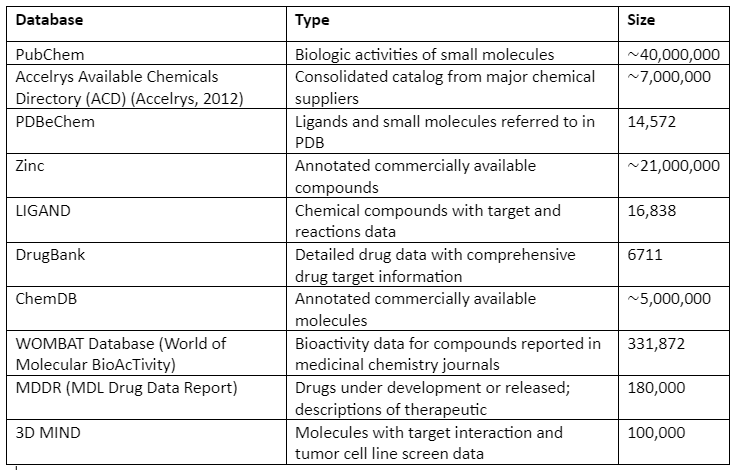
In addition to general libraries, there are focused libraries that as the name indicates are designed specifically for a family of related targets. The last one is the targeted libraries that are designed specifically for a single target.
Structure-Based CADD
Structure-Based computer-aided drug design (SB-CADD) depends upon the capability to predict and analyze the 3D structure of many biological molecules. The core principle or hypothesis of this technique is that a molecule can interact with a specific type of protein and this interaction produces a biological effect that depends upon the ability of a molecule to favorably interact with a specific binding site present on that protein. These favorable interactions shared by molecules will have a similar biological effect. Thus, we can identify novel compounds by analyzing the binding site of the protein. Also, for any SB-CADD, structural information of the target is required.
The steps involved in SB-CADD are given by:
Step one: Determination of Target Structure
As mentioned, target structure is a prerequisite for any SB-CADD. The structure of the target molecule is determined through techniques like NMR or X-ray crystallography and then it is submitted in protein databank (PDB). The rate of structure determination has been enhanced by using structural genomics techniques. This has allowed to carry out some successful vHTS even in the absence of experimentally-determined structures. Besides this, for any successful screening process quality is structural information of target protein and the small molecule or compound is necessary. The first step in drug design is to identify whether the target protein has suitable binding pockets or not. This step is accomplished by using in silico methods for the target-ligand analysis of the structure to find potential binding affinities. Some of the methods are:
Comparative Modeling:
It determines the target structure by using a template having a similar sequence, based on the assumption that the structure of a protein is more conserved than the sequences, in other words, similar sequence proteins have the same structural features. Among comparative modeling, homology modeling is the widely used type. In homology modeling, the template and the target proteins have the same evolutionary origin.
The steps of comparative modeling are:
Identification of template protein
Sequence alignment of the template and target protein
Coordinated copying for perfectly aligned regions
Building missing atom coordinates of target protein structure
Refinement of model and evaluation
For automated comparative modeling, many programs exist, two widely used are PSIPRED and MODELER. Also, important model databases are SWISS-MODEL and MODEBASE. The comparative modeling is illustrated by the following figure:
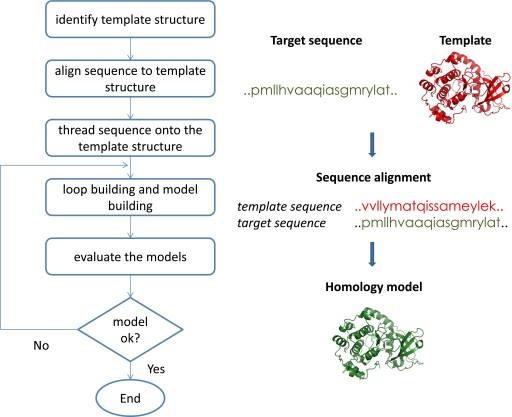
Overview of Comparative Modeling
Step Two: Detection of Binding sites and Characterization
After the structure of the target protein is known, it is important to elucidate protein-ligand binding interaction which is a necessity for drug discovery. This is done through the identification of binding sites on the target protein. Mostly the binding sites are identified from cocrystal structures of target or through closely related proteins having natural or synthetic ligands. In the cases where cocrystal structure is not present, mutational studies help to identify the binding sites. Also, for the identification of new binding sites or unknown binding sites, computational tools like Q-SITEFINDER, POCKET, SURFNET, etc. are used.
The computation for identification and characterization of binding sites can be divided into three types:
Geometric methods: these methods determine the binding site on the target by detecting cavities present on the protein surface. They use grids to describe the 3D structure of the protein. The pocket boundary is determined by rolling a “spherical probe” over the surface of the grid and a pocket is determined if there is a period of non-interaction that is probe after rolling does not touch any target molecule. this technique is employed by POCKET, LIGSITE, and SURFNET.
Energy-based Methods: these methods determine the favorable binding affinities by taking into account Van der Walls, electrostatic, hydrogen-bonding, solvent, and hydrophobic interactions of the probe. These simple methods are efficient and sensitive as compared to the geometric methods. The tools that employ these methods include Q-SITEFINDER, GRID and POCKETPICKER.
Molecular structure dynamics-based methods: As we all know, the structure of biomolecules is diverse and dynamic that cannot be depicted through a single static structure predicting putative binding sites. In these approaches, multiple conformations of target molecules are taken into account for determining the binding sites of the target. These include classical molecular dynamics (MD) simulations for getting an ensemble of target protein conformations starting from a single structure.
Step Three: In silico representation of Target and Ligand Structure
There are three types of methods to depict the structures of target and ligand. These methods are given by:
Atomic representation: This is commonly used when ranking and scoring are based on potential energy function. For instance, DARWIN used the CHARMM force field for the calculation of energy.
Surface representation: these methods describe the topography of molecules with the help of geometric characteristics. The surface is shown as a network of smooth convex, concave, and saddle shape surfaces. These are mapped with the help of van der Wall forces by making use of a sphere probe. In addition, docking is then done by complementary alignment of target binding surface and ligand. For example, DOCK.
Grid representation: In this type of representation, the target protein is encoded as a physiochemical feature of its surface. Katchalskikatzir and his colleagues have digitalized molecules by employing a 3D discrete function that discriminates the surface from inside of the target molecule. After that, molecules are scanned in three dimensions in relative orientation, and then the extent of overlap is determined between molecules with the help of a correlation function which is calculated from a Fourier transform. From the list of overlaps, the best one is selected. Also, the grid shows physiochemical properties by storing energy potentials on surface grid points.
Step Four: Sampling Algorithm for Protein-Ligand Docking
The methods of docking can be categorized as rigid-body docking and flexible docking. This classification depends on the extent to which the flexibility of ligand and target is considered during docking. In rigid-body docking, only static geometric/physiochemical complementarities are considered between target and ligand. It ignores flexibility and induced-fit models. This docking method is preferred where a large number of compounds need to be docked in a short time. Besides this, for docking, flexible methods are necessary. But these flexible methods are still under the phase of optimization and refinement as there are several possible conformations of target and ligand. Some of the widely used methodologies include systematic enumeration of conformations, Monte Carlo search algorithms with Metropolis criterion (MCM), genetic algorithms, and molecular dynamic simulations.
Step Five: Structure-Based Virtual High-Throughput Screening
Virtual high-throughput screening is a very important step in CBDD. It is the main step that differentiates between normal drug discovery and computational drug discovery. SB- vHTS identifies “hits” out of hundreds of compounds to a target whose structure is known. The identification depends upon the 3D structure comparison of a small molecule with the target molecule binding pockets. This method made the screening of hundreds of compounds possible in a short time as compared to traditional screening in which the binding affinity was assessed experimentally.
To make virtual screening of a large number of compounds feasible, SB-vHTS utilizes limited conformational sampling of ligand and protein, and also it makes use of a simplified approximation of binding energy that can be computed quickly. This approximation can also result in the false-positive result but it can be removed by selecting the best-ranked molecules having the best binding affinities and also making use of iterative docking and clustering ligand positions. Important steps involved In SB-vHTS are:
Library preparation for docking of target protein and compound
Determination of favorable binding positions for both target and ligand
Ranking of docked structures
Step Six: Scoring Functions for Analysis of Protein-Ligand complexes
The accurate assessment of protein-ligand complexes after the application of docking is a crucial step. After the ligand docking, there are hundreds and thousands of possibilities of target-ligand complex interaction and to have the best analysis, a scoring function is required. The scoring function will help in ranking these complexes and also it will be able to differentiate between valid binding mode predictions from invalid predictions. In addition to this, highly complex scoring functions also attempt to predict target-ligand binding affinities for hit-to-lead and lead-to drug optimization.
The scoring functions can be grouped into four categories:
Force-Field scoring functions
Knowledge-based scoring functions
Empirical scoring functions
Consensus scoring functions
After the hit is identified, then it is tested in vitro to obtain the potential drug. Thus, SB-CADD allows us to identify novel compounds for potential drugs, and also it identifies new binding sites at an efficient pace.
Ligand-Based CADD
The ligand-based computer-aided drug discovery (LB-CADD) is based on the analysis of known ligands to interact with the target protein. In these methods, a group of reference structures is collected from compounds that have been already known to interact with the target protein, and then their 2D and 3D structures are analyzed. The overall aim is to show these compounds in a way that the physiochemical properties which are needed for desired interactions are retained and the other information which is not required for interaction is eradicated. This approach is assumed to be an indirect method of drug discovery as this method does not need prior knowledge of the target of interest.
The LB-CADD has two approaches:
Compound selection based on chemical similarity to known active compounds by employing some sort of similarity measure
Development of a quantitative structure-activity relationships (QSAR) model which envisages the biological activity from a chemical structure.
The difference between the two approaches lies in the fact that in the second method, features of the chemical structures are taken into account having an appropriate biological activity while nothing of such sort is assumed in the first method. Both of the methods are used to find in silico novel potential drugs which have biological activity and also can optimize DMPK/ADMET properties.
It is stated that LB-CADD is based on the Similar Property Principle, which states that the molecule which has the same structural features are likely to have the same properties. Thus, these approaches can identify a potential ligand even when the target protein structure is not known. Also, the ligands identified through this approach are more potent than the SB-CADD. Two important approaches employed in LB-CADD are explained below:
Pharmacophore modeling
Pharmacophore is composed of a molecular framework that describes the important characteristics that are responsible for the biological activity of a compound. When the structure information of the target is not known then pharmacophore models are developed by using structural features of active ligand molecules that can bind to the target protein. In other case scenarios, where the structure of the target molecule is known then, the binding site information can be utilized to built pharmacophore models. The pharmacophore models employing chemical properties like acidic/basic residues and H-bond donor or acceptor are determined to be the most potent models. In addition to this, pharmacophore modeling is also used for the virtual screening of drugs present in large databases. The programs that generate and identify pharmacophore models are DISCO, GASP, and Catalyst.
The pharmacophore model construction has the following steps:
Identification of active ligand compounds that are known to bind with the target either by literature search or through the database.
For the 2D model, important types of atoms and their connectivity are defined while for the 3D model the conformations are defined by using IUPAC nomenclature.
Alignment of ligand or superimposition is employed to find the common properties needed in binders.
Building of pharmacophore model
Ranking of pharmacophore model and selection of the best model.
In vitro validation of pharmacophore models.
QSAR (quantitative structure-activity relationships)
These methods are based on statistics that associate or compare activities of target drug interactions with different molecular descriptors. The main principle of QASR is that the molecule having the same structure shows similar biological features and activity. These models elucidate mathematically how the activity of a target will change with the binding of a different ligand. So, QASR is calculated by measuring the correlation between experimental biological activity and many properties of small ligand binders. This QASR relationship can also be used to guess the new drug molecule analogs’ activity.
The important steps involved in QASR are:
Identification of active ligand compounds and their activity that are known to bind with the target either by literature search or through the database
Identification of physicochemical or structural molecular features that affect the biological activity (e.g., functional group, surface area, bonding, etc.)
Development of QASR between biological activity and identified property of the drug molecule.
Authentication of QASR biological activity predictive power.
Optimization of the known active compound by using the QSAR model to maximize its biological activity
The testing of new optimized drug
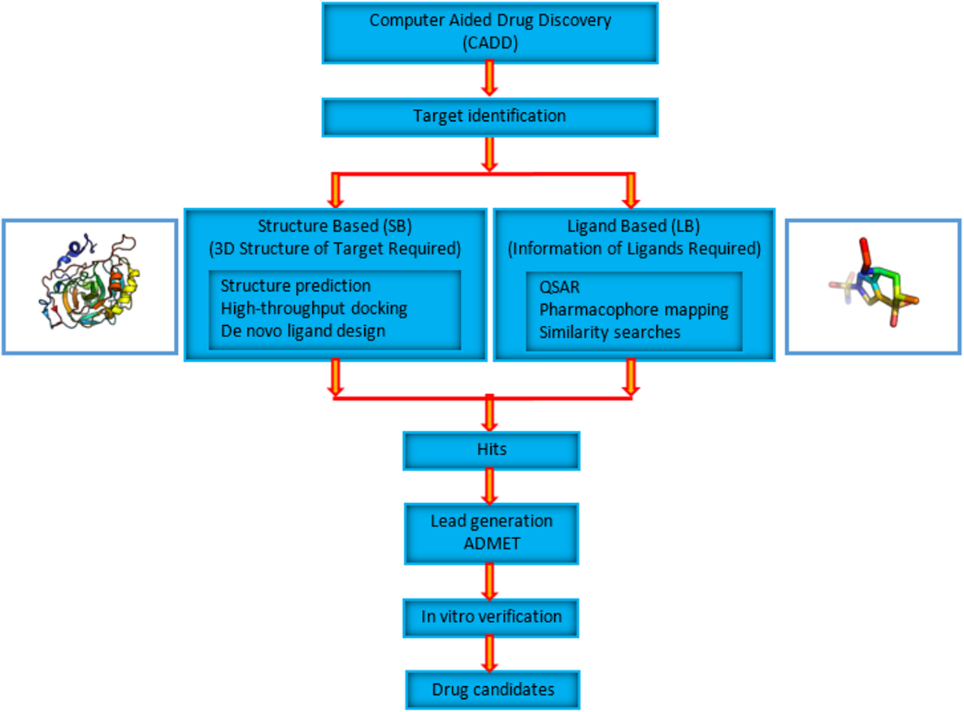
CADD Pipeline
Conclusion
No doubt, CADD has now been extensively used for drug discovery but still many important therapeutic targets like protein-protein interactions and protein-DNA interactions are still a problem as they have large sizes making them complex. Also, computational tools operations required a trained person to handle them as some of the tools are not quite user-friendly, making their accessibility a problem. Still, works need to be done to make CADD more accessible and efficient.
