We provide complete services for Computational Drug Discovery Services
From data collection, preprocessing, quality control to transcription factor binding sites, histone modifications and much more.
Providing services to
worldwide clients
Datasets
analyzed
Qualified researchers and
Bioinformatics analysts
Satisfied orders processed
Our Services
- Target Identification Services
- HIT Discovery Services
- Lead Optimization Services
- 3D Structure Prediction Services
- Molecular Docking Services
- ADMET Analysis Services
- Molecular Dynamics Simulation Services
- Drug Repositioning Services
- Structure-based Drug Design Services
- Ligand-based Drug Design Services
- Virtual Screening Services
- Antibody Designing
- Complete end-to-end computational drug discovery services
- Complete end-to-end computational peptide docking and design services

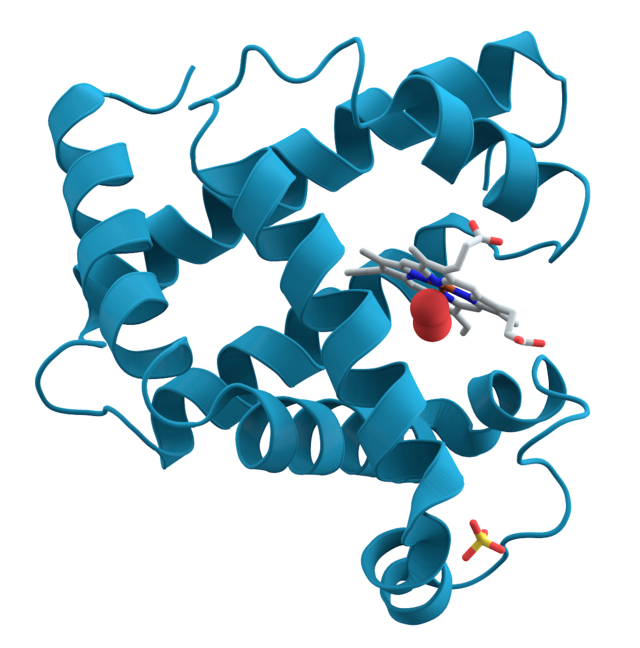

Computational Drug Designing has become the go-to requirement for the researchers, scientists and the pharmaceuticals who fight against the fatal disease. Computational drug discovery is an effective strategy for accelerating and economizing drug discovery and development processes. Because of the dramatic increase in the availability of biological macromolecule and small molecule information, the applicability of computational drug discovery has been extended and broadly applied to nearly every stage in the drug discovery and development workflow, including target identification and validation, lead discovery and optimization and preclinical tests.
Bioinformatics analysis can not only accelerate drug target identification and drug candidate screening and refinement, but also facilitate characterization of side effects and predict drug resistance. High-throughput data such as genomic, epigenetic, genome architecture, cistromic, transcriptomic, proteomic, and ribosome profiling data have all made significant contributions to mechanism-based drug discovery and drug repurposing. Accumulation of protein and RNA structures, as well as development of homology modeling and protein structure simulation, coupled with large structure databases of small molecules and metabolites, paved the way for more realistic protein-ligand docking experiments and more informative virtual screening.
During the early stages of drug discovery for a certain disease, the underlying molecular mechanisms behind the disease are studied. These studies include identifying the cellular and genetic factors involved in the disease, followed by the identification of potential targets. In order to ensure that the biological target is involved in the disease, in vitro (isolated cells) and in vivo (animal models) tests are performed. This is also known as the target validation stage. Modern target validation often involves a combination of in vitro, in vivo, and in silico (performed with a computer) studies. The results of the target validation stage can assist in lead compound identification. Lead compounds are chemical compounds that show desired biological or pharmacological activity and may initiate the development of a new clinically relevant compound. Lead compounds are typically used as starting points in drug design to give new drug entities. Drug design strategies can be used to improve the compound’s pharmacodynamic and pharmacokinetic properties.
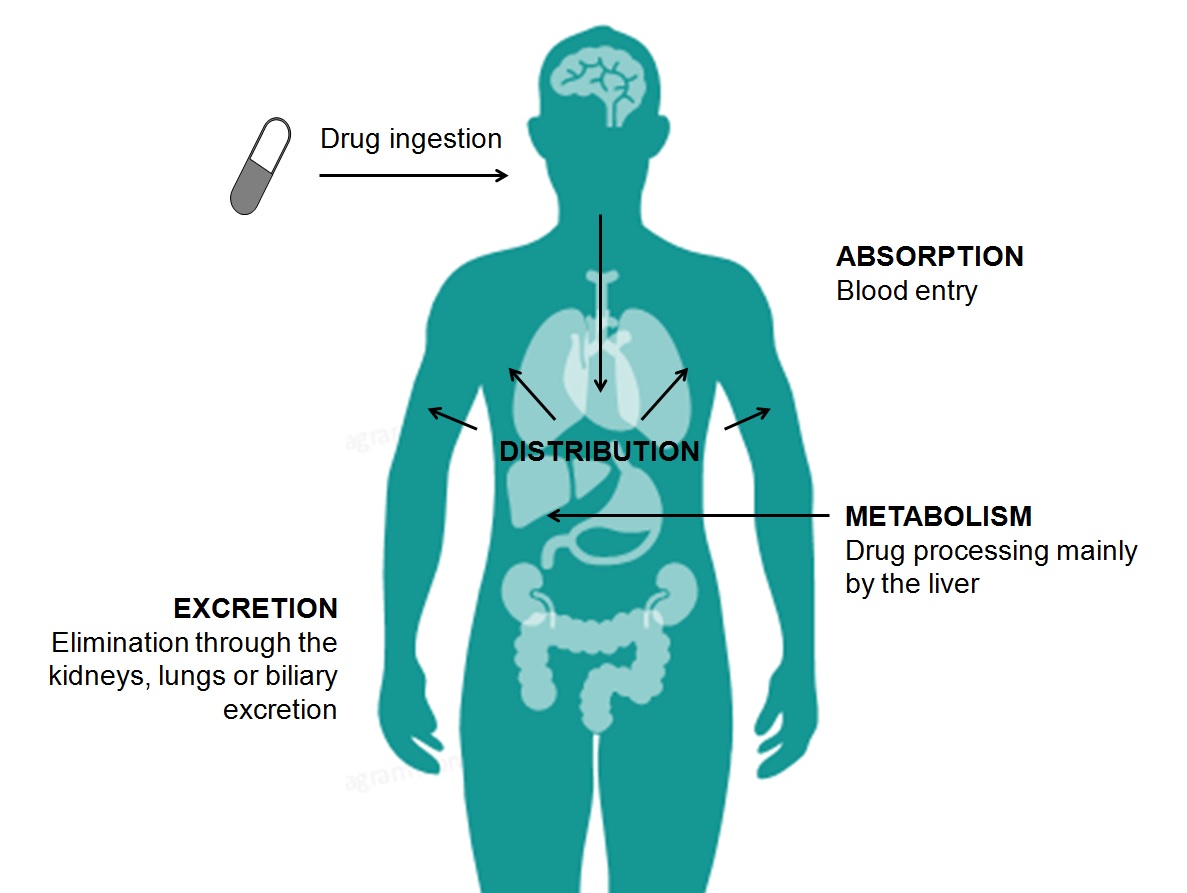
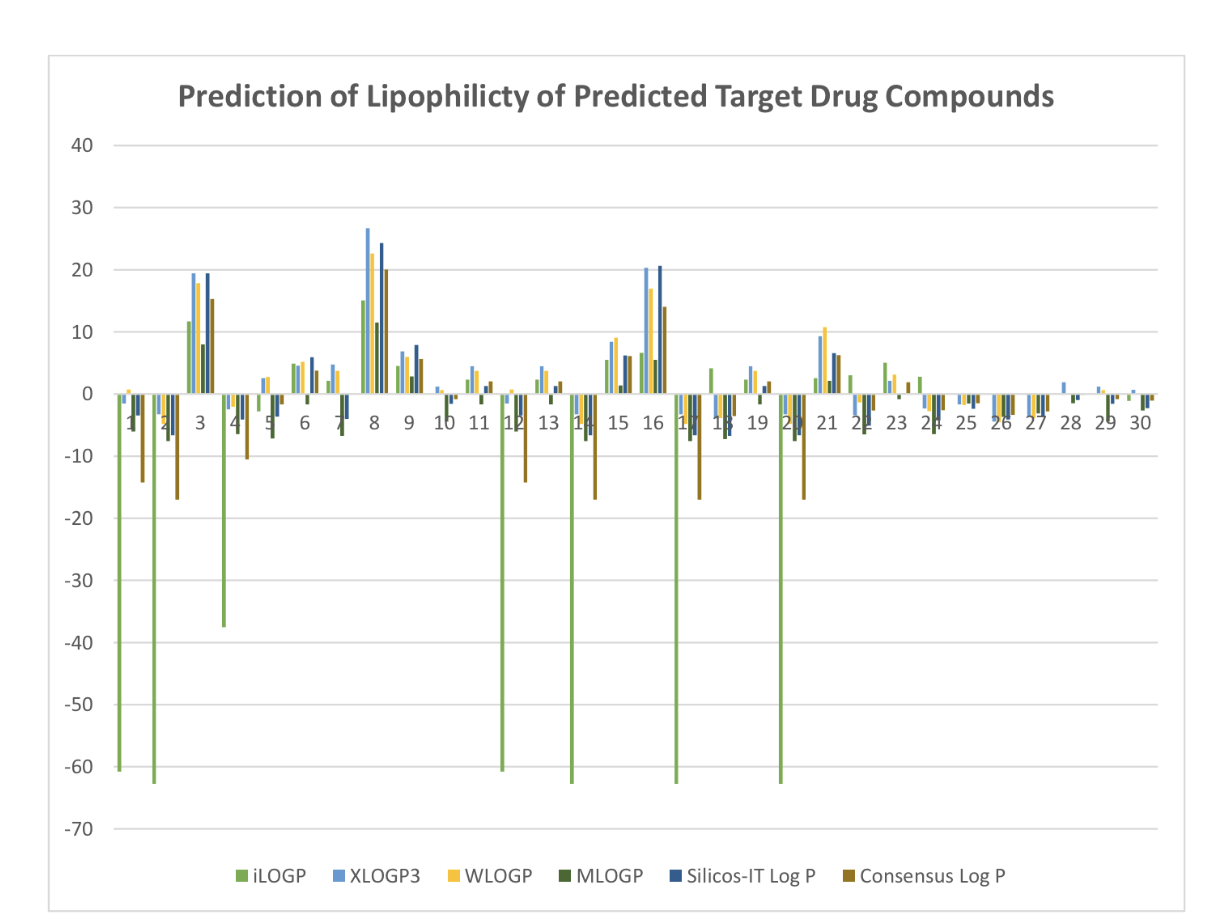
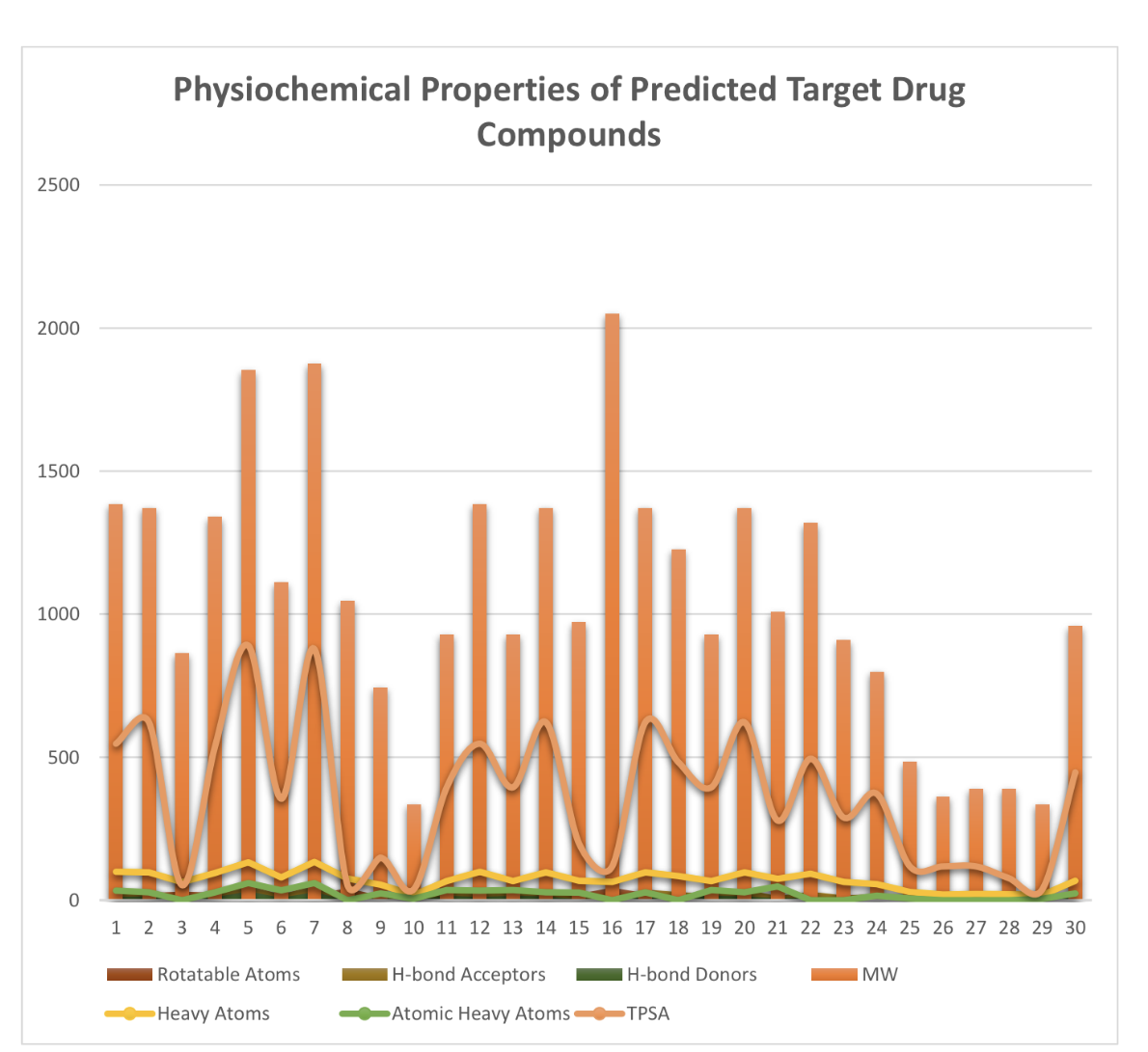
ADMET Properties
ADMET stands for chemical absorption, distribution, metabolism, excretion, and toxicity. These properties play crucial roles in drug discovery and development. A high-quality drug candidate should not only have sufficient efficacy against the therapeutic target, but also show appropriate ADMET properties at a therapeutic dose. A lot of in silico models are hence developed for prediction of chemical ADMET properties. However, it is still not easy to evaluate the drug-likeness of compounds in terms of so many ADMET properties.
Protein 3D Structure Prediction
Protein tertiary structure known as 3D protein structure is the three dimensional shape of a protein. The tertiary structure will have a single polypeptide chain “backbone” with one or more protein secondary structures, the protein domains. Amino acid side chains may interact and bond in a number of ways. Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence.
Homology Modeling
Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the target protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the template). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence.
Servers for Homology Modeling:
SWISS-MODEL, AWSEM-Suite, ROBETTA, FoldX, MODELLER, MOE and many others.
Fold Recognition/Threading
Protein threading/fold recognition is a method of protein modeling which is used to model those proteins which have the same fold as proteins of known structures but do not have homologous proteins with known structure. It differs from the homology modeling method of structure prediction as protein threading is used for proteins which do not have their homologous protein structures deposited in the Protein Data Bank (PDB) whereas homology modeling is used for those proteins which do. Threading works by using statistical knowledge of the relationship between the structures deposited in the PDB and the sequence of the protein which one wishes to model.
Servers for Protein Threading:
IntFOLD, HHpred, RaptorX, Phyre and Phyre2 etc.
Ab Initio/De Novo Structure Prediction
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure. De novo protein design involves the production of novel protein sequences that adopt a desired fold.
Servers for Ab Initio Structure Prediction:
I-TASSER, trRosetta, Abalone, ROBETTA and others.
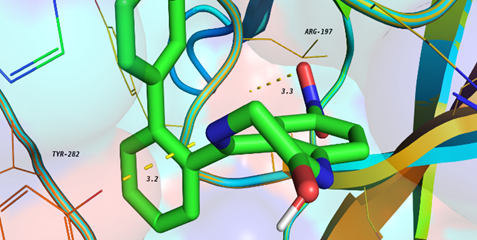
Molecular Docking
Molecular Docking is a bioinformatics modeling technique which involves the interaction of two or more molecules to give the stable adduct. Molecular docking is an important approach for designing new drugs and vaccines and other bioinformatics analysis as well. It predicts the three-dimensional structure of any complex depending upon binding properties of ligand and target.
Molecular interactions including protein-protein, enzyme-substrate, protein-nucleic acid, drug-protein, and drug-nucleic acid play important roles in many essential biological processes, such as signal transduction, transport, cell regulation, gene expression control, enzyme inhibition, antibody–antigen recognition and even the assembly of multi-domain proteins. These interactions very often lead to the formation of stable protein–protein or protein-ligand complexes that are essential to perform their biological functions.
Tools and Servers available for Molecular Docking
AutoDock, SwissDock, rDock, PatchDock, VirtualFlow, MOE (Molecular Operating Environment) and many others.
Molecular Dynamics Simulations
Molecular Dynamics (MD) is a computer simulation approach permitting the time evolution prediction of an interacting particular system which involves the generation of atomic trajectories of a system using numerical integration of Newton’s laws of motion to define specific interatomic potential using the initial condition and boundary condition.
MD simulation analysis is one of the essential steps while designing novel drugs using computational approaches. MD analysis not just evaluates and validates the protein structures, either predicted computationally or experimentally, but also validates the drug-target compatibility by providing various statistical information about the interacting drug-target complex.
Peptide Designing
Peptides are complex biomolecules that have unique chemical and physical properties that are a direct result of their amino acid composition. The development of peptide drugs requires specific considerations of this family of biopolymers. Historically, peptide vaccines to viral infections and antibacterial peptides led the way in clinical development, but recently many other diseases have been targeted, including the big sellers AIDS, cancer, and Alzheimer’s disease. Synthetic peptides are used in polypeptide structure/function studies, antibody production and as peptide hormones or hormone analogues. They are also used to design novel enzymes, drugs and vaccines. Tools that are used for peptide designing include Peptide-Mine, EPI-peptide designer etc.
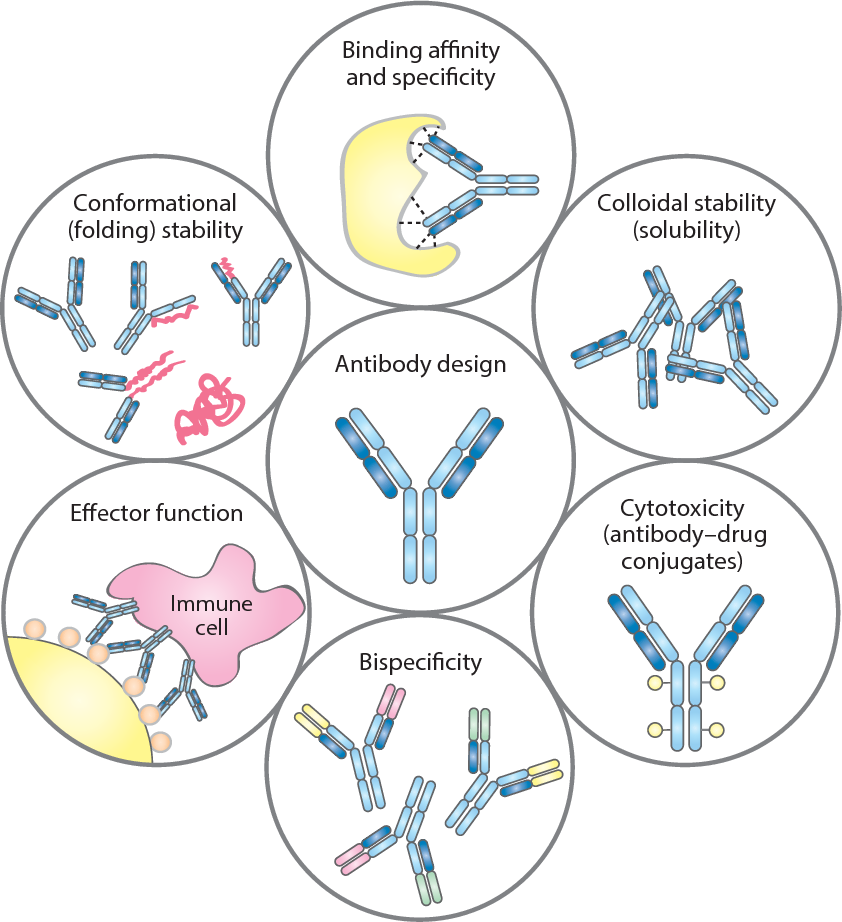
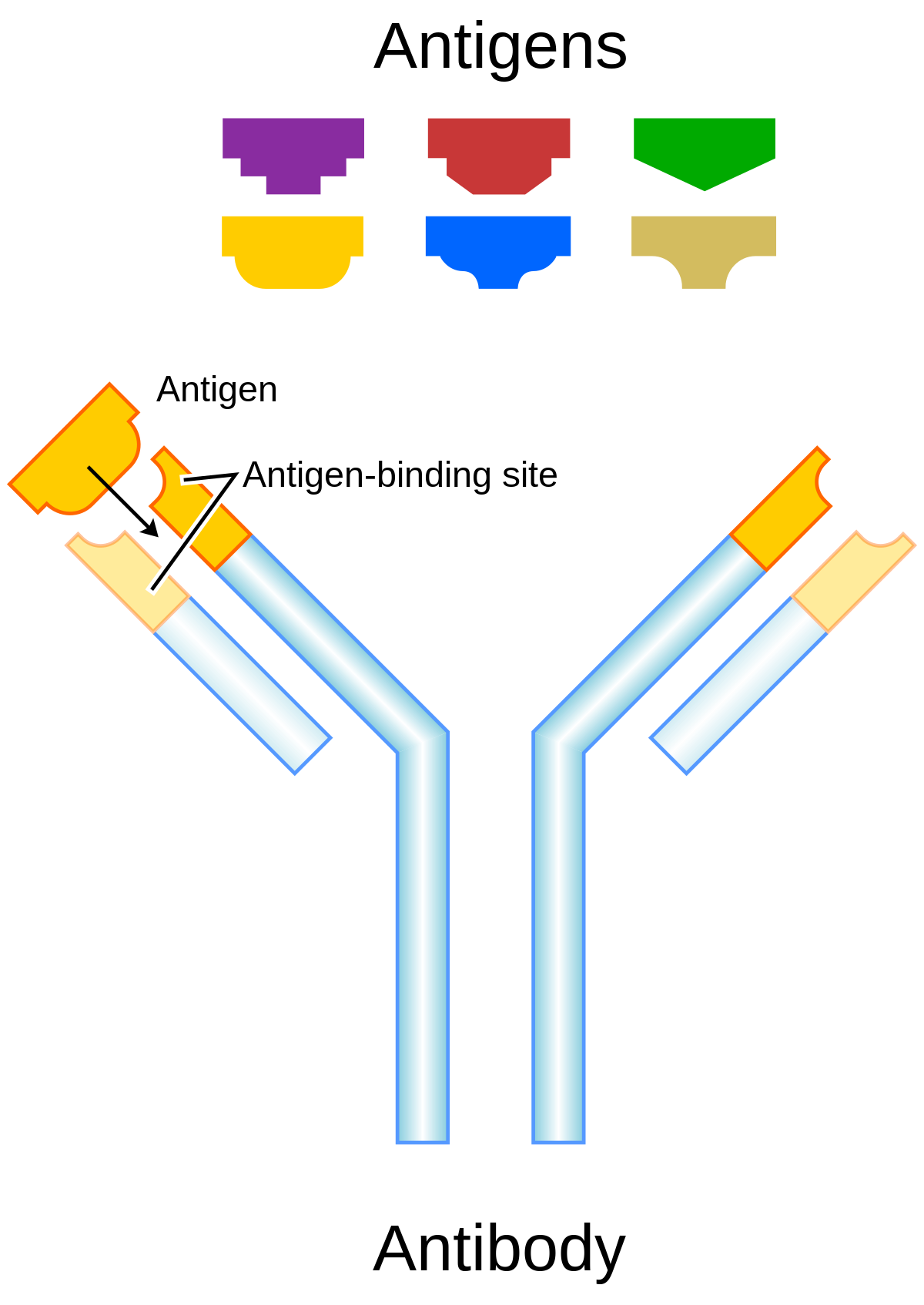
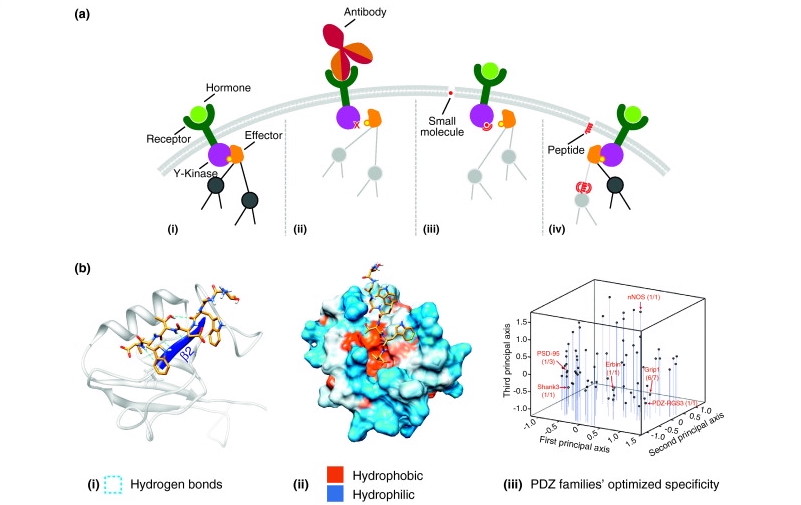
Antibody Designing
Antibodies are immune system proteins that recognize the surfaces of foreign molecules (antigens) for subsequent elimination from the organism during an adaptive immune response or self-antigens from healthy tissues in autoimmune diseases. Computational advances in protein modelling and design can have a tangible impact on antibody-based therapeutic development. Antibody-specific computational protocols currently benefit from an increasing volume of data provided by next generation sequencing and application to related drug modalities based on traditional antibodies, such as nanobodies. Different tools are used to design antibodies for instance AbDesign, BioPhi etc.
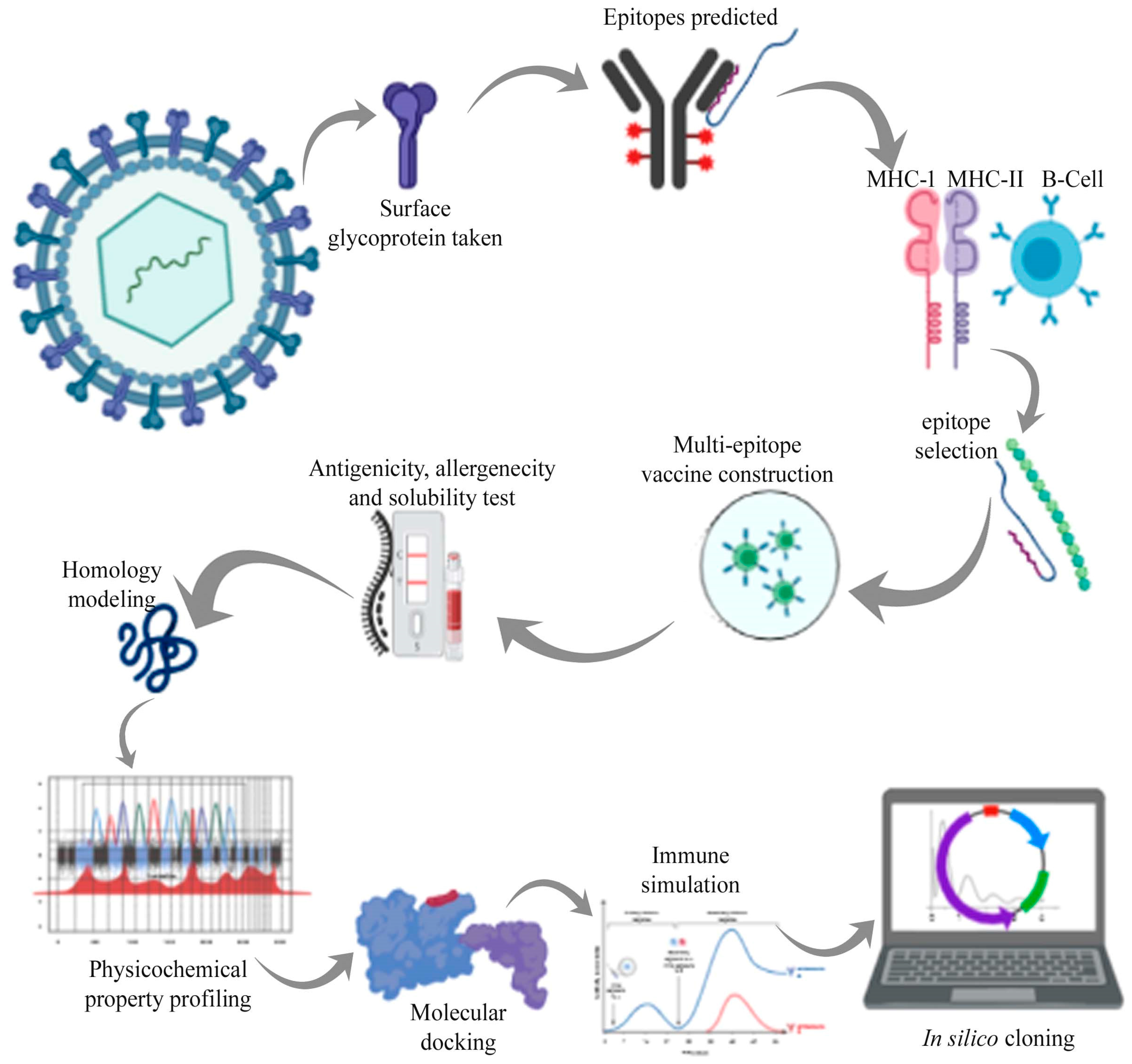
Reverse (Computational) Vaccinology and Immunoinformatics
Reverse vaccinology is the state of the art vaccine development strategy that starts with predicting vaccine antigens by bioinformatics analysis of the whole genome of a pathogen of interest. Reverse vaccinology defines the process of antigen discovery starting from genome information.
Immunoinformatics is a field of Bioinformatics and Immunology combined that helps in identification of key role playing factors in a particular disease and immune response condition, and then design therapeutics for such diseases and conditions. Immunoinformatics deals with computational techniques and resources used to study the immune functions.
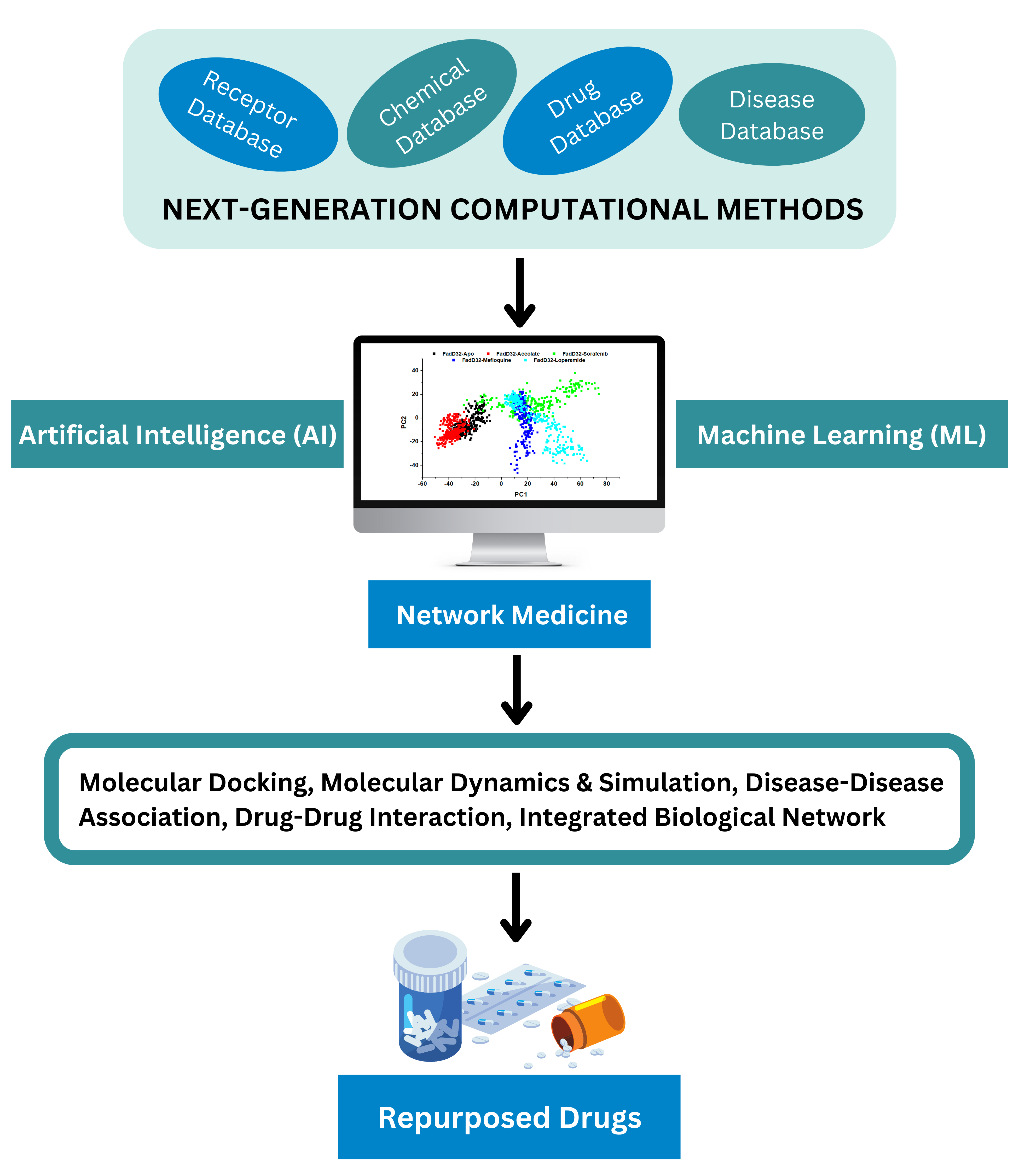
Drug Repurposing
Drug repurposing or drug repositioning is a process of identifying new therapeutic uses for the existing available drugs. It is an effective strategy in discovering or developing drug molecules with new pharmacological/therapeutic indications. Drug repurposing involves exploring new medical uses for existing drugs including approved, discontinued, shelved and investigational therapeutics.
Repurposing of old drugs to treat both common and rare diseases is becoming attractive because of high attrition rates, substantial costs and slow pace of new drug discovery. Repurposing involves the use of de-risked compounds with potentially lower overall development costs and shorter development timelines.
Get in touch with us and we will start your analysis right away!
