We provide complete services for Computational Vaccine Designing and Immunoinformatics Services
From data collection, preprocessing, quality control to transcription factor binding sites, histone modifications and much more.
Providing services to
worldwide clients
Datasets
analyzed
Qualified researchers and
Bioinformatics analysts
Satisfied orders processed
Our Services
- Chimeric Vaccines
- Multi-Chimeric Vaccines
- Multi-Epitope Vaccines
- Reverse Vaccinology
- Peptide Based Vaccines
- Structure-based Drug Design Services
- Fragment-based Drug Design Services
- De novo Design Services
- Multiple Targeting Design Services
- Pharmacophore Model Construction Services
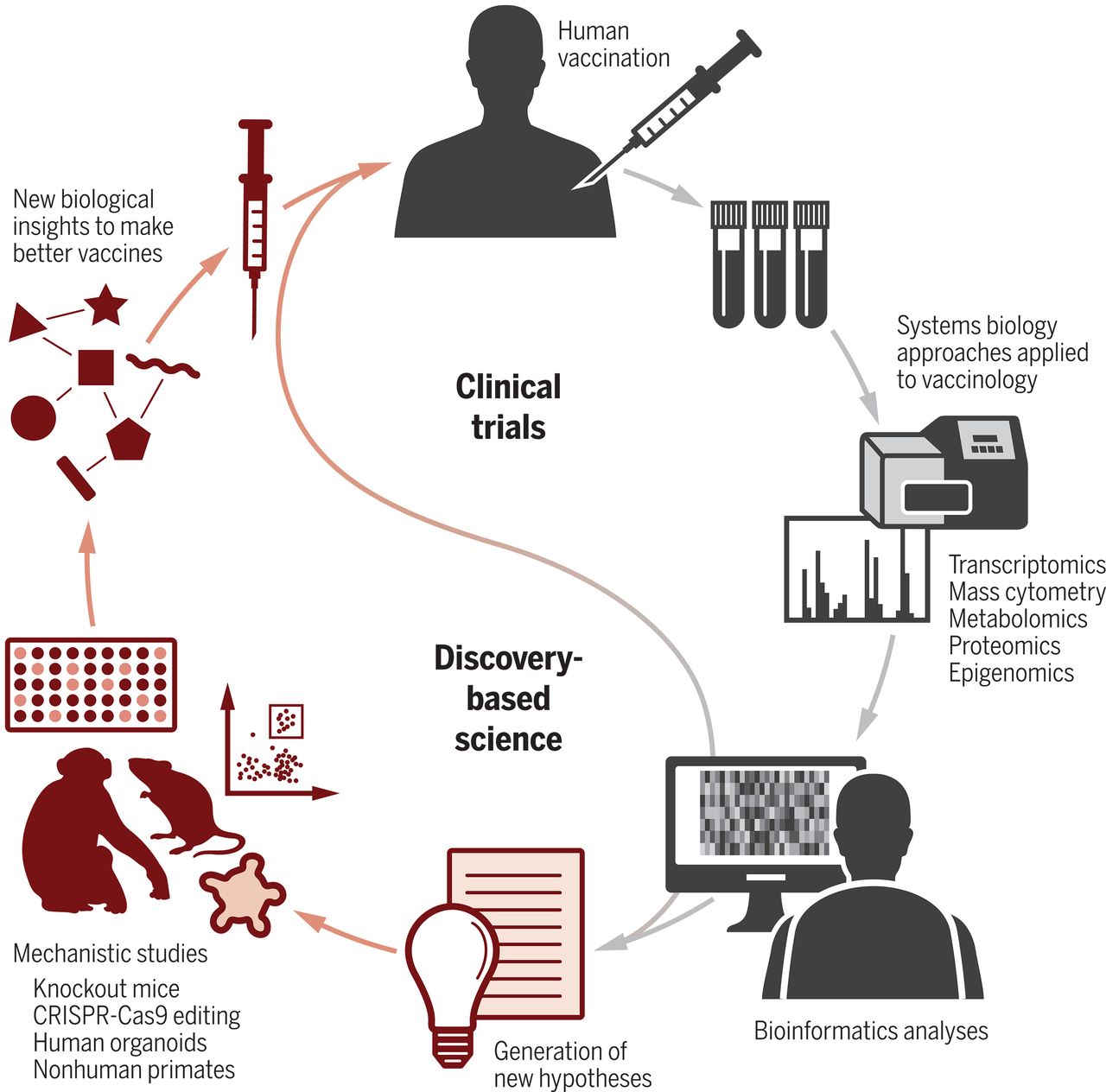
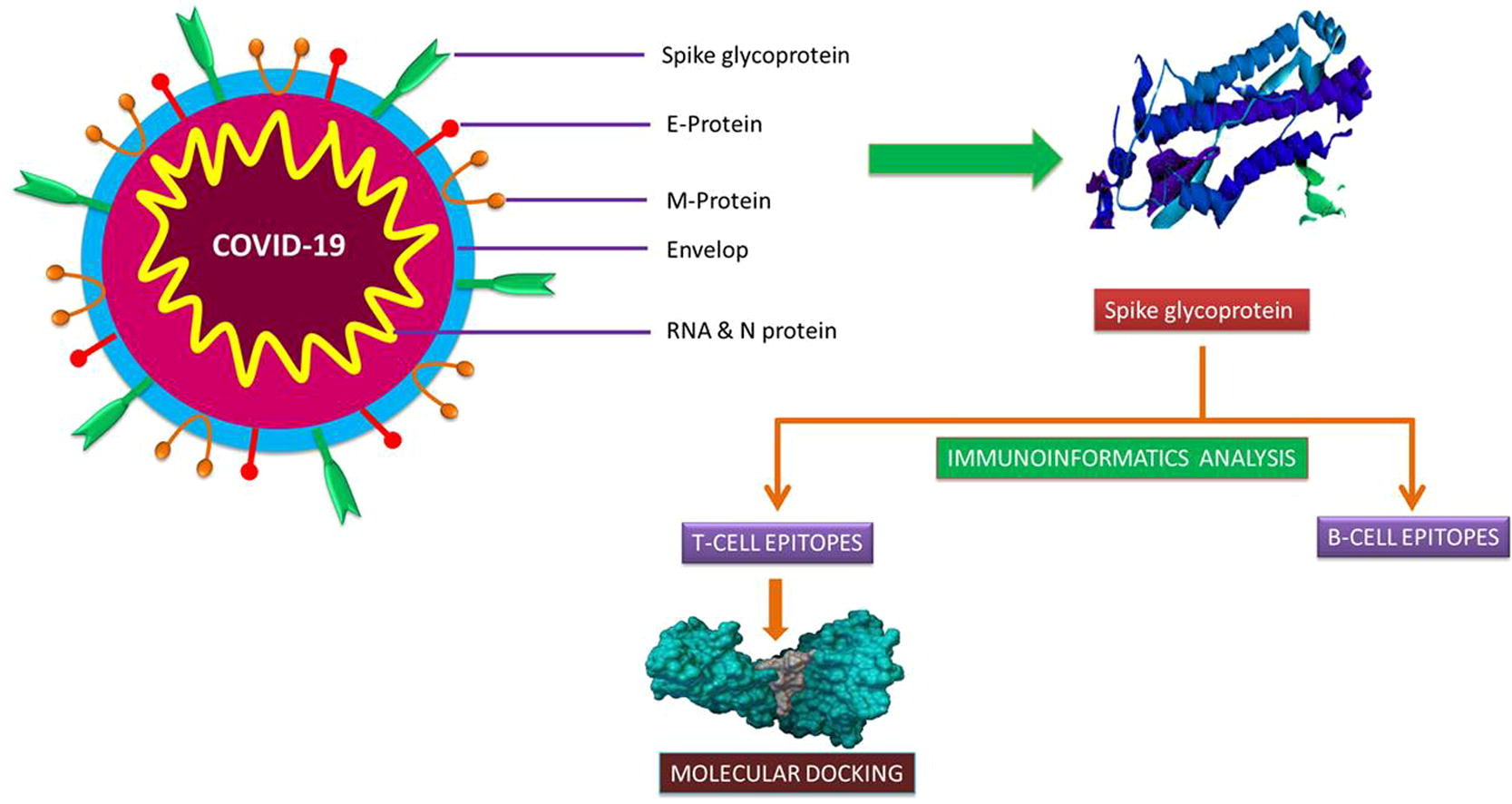
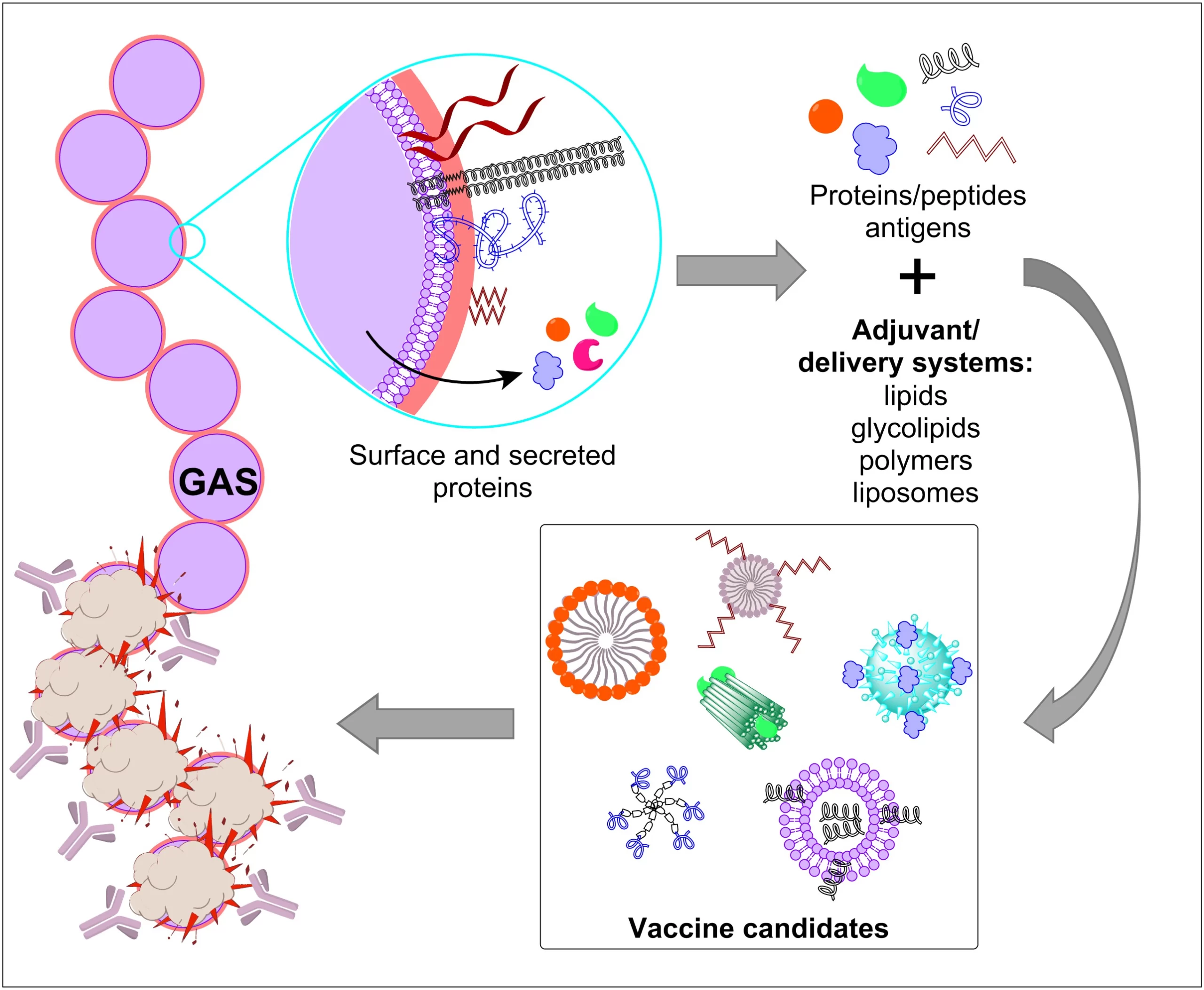
Newly emerging and reemerging infectious viral diseases have threatened humanity throughout history. The unprecedented scale and rapidity of dissemination of recent emerging infectious diseases pose new challenges for vaccine developers, regulators, health authorities and political constituencies. Vaccines are biological substances that are utilized to stimulate antibody production within the body of an organism to provide active immunity against foreign organisms, mostly viruses and bacteria. Vaccines not only arrest the beginning of different diseases but also assign a gateway for its elimination and reduce toxicity. Vaccines are the most cost-effective public health interventions.
Bioinformatics is also involved in medication development aimed at bio-productive and pharmaceutical/vaccine development. Computational approach in drug discovery helps in identifying safe and novel vaccines. In silico(computational) analysis saves time, cost, and labor for developing the vaccine and drugs. Chimeric vaccines are types of recombinant vaccines, produced by substituting genes from the target pathogen in a closely related organism, for similar genes. Chimeric vaccines are useful in studying infectious diseases, including many neglected diseases.
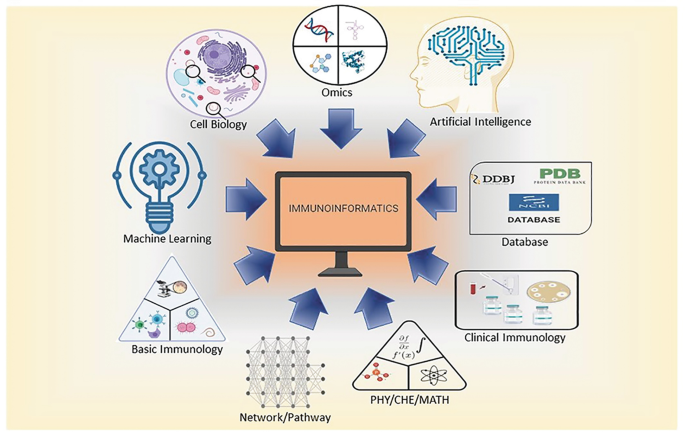
Immunoinformatics, otherwise known as computational immunology, is the interface between computer science and experimental immunology. It represents the use of computational methods and resources for the understanding of immunological information. It not only helps in dealing with huge amounts of data but also plays a great role in defining new hypotheses related to immune responses. Immuno-Bioinformatics is being extensively used in designing B-, T-cell epitopes, vaccines, antibodies, adjuvants, diagnostic kits, and therapeutics.
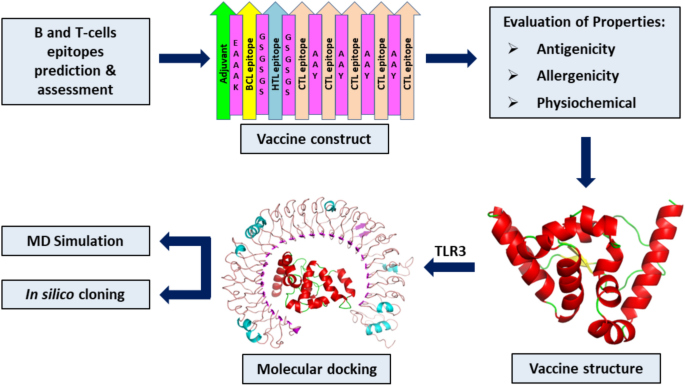
Computational Vaccine Designing Process
In order to design a vaccine computationally the followings steps are performed:
Target Identification
In vaccine development the first step is to identify the target in order to start the process of computational construction of vaccines. Antigen identification is an important step in the vaccine development process. Vaccine targets are selected from conserved regions in the genome of the pathogen (organism that causes disease) in question, with the aim of conferring broad and lasting immunity.
Following steps are performed during target identification:
Select the target for the vaccine
Selection of the target is a primary and tough step for target identification in vaccine development. The vaccine target should be any unique part of the microorganism which could be enabled to introduce a protective immune response in the host. The UniProt database can be used for the retrieval of the complete proteome of the pathogen .
Remove duplicates from the retrieved biological data
All the paralogous sequences from the proteome can be removed by using computational bioinformatics well-known suite called “CD-HIT”. It is an acronym of Clusters Database at High Identity with Tolerance mostly used by the biotechnologists and bioinformaticians to remove continuous repetitive part of the sequences within the required genome. It takes the sequence in FASTA format and gives the files in compressed form containing non-repetitive and non-redundant protein sequences that are totally free from paralogs as output.
Screen Non-homologous Proteins
Non-homologous proteins are screened by using BLASTP, which is an online accessible tool used by many biologists. It basically compares primarily biological sequences of proteins against protein databases and generates their sequence alignments. Protein database used mostly for screening the non-homologous proteins is RefSeq, which is a well-annotated, non-redundant, non-repetitive sequence database. Screening the non-homologous proteins is important as in this task, all the proteins that are present both in host and pathogen are eliminated and only the non-homologous proteins are kept in record.
Immunoinformatics Approach for Epitope Prediction
The rapid development of secure, accurate, easy, cost-effective, trustworthy, and in silico development of epitope-based vaccines against viruses allows for the fast establishment of innate immunity against the directed antigen. Immunoinformatics approaches are both cost-effective and convenient.
For epitope prediction we will perform the following steps:
Screen Antigenicity of Protein
Vaccine development needs target specific antibodies that are pivotal for the immunodiagnostic tests, identification of protein-DNA, studies on proteomics for the discovery of cancer biomarker, and other interactions, and small and large biochemical assays. Therefore, it is important to screen the properties of protein sequences that are important for antigenicity and to identify small peptide epitopes and large regions in the linear sequence of the proteins whose use results in specific antibodies.
Predict Linear B-Cell Epitopes
B-cell epitope prediction helps to facilitate the identification of B-cell epitope with the practical purpose of replacing the antigen for antibody production or for carrying structure-function studies or in vaccine development. B-cells can use pathogenic molecules by targeting them with high and unique specificity by using receptors present on their surface. B-cell epitopes are rich in polar residues containing charges, loops and depleted in helix to beta strands. They are a key source in our adaptive immunity. ABCPred server is most useful in predicting the B-cell epitopes areas in an antigen sequence that are very useful in searching synthetic vaccine candidates, disease diagnosis and also in allergy research.
Assess Linear B-Cell Epitopes
Assessment helps the researchers to predict whether the B-cell epitopes of proteins are antigenic or not. In this assessment the values for antigenicity are checked. This assessment can be done by using a well-known server “Immune Epitope Database Analysis Resource”. This server gives us a collection of tools for the prediction and analysis of immune epitopes.
Predict CTL Epitopes
Cytotoxic T lymphocyte (CTL) are potential epitopes candidates for subunit vaccine design for many diseases. CTL epitopes recognize particular antigens. Indirect methods are used for the prediction of CTL epitopes. These methods are based on the prediction of MHC class I binders instead of CTL epitopes. This prediction can be done by using a tool “MHC-I Binding Predictions” available at well-known server “Immune Epitope Database Analysis Resource”.
Computational Construction of the Vaccine
In computational construction of the vaccine firstly assessment and prediction are performed which are necessary for the construction of the vaccine.
Assess CTL Epitope
Indirect methods are used for the prediction of CTL epitopes. This assessment can be done by using a well-known server “Immune Epitope Database Analysis Resource”. This server gives us a collection of tools for the prediction and analysis of immune epitopes. Along this, to check the antigenicity of epitopes use the “Vexigen ver2.0” server.
Predict and Assess HTL Epitope
The responses of Helper T lymphocyte (HTL) play an important role in the induction of both humoral and cellular immune responses. This assessment can be done by using a well-known tool “MHC-II Binding Predictions” available at “Immune Epitope Database Analysis Resource” server. This server gives us a collection of tools for the prediction and analysis of immune epitopes CTL, HTL epitopes etc. To check the antigenicity of HTL epitopes use the “Vexigen ver2.0” server.
Map Vaccine Construct
To construct the final multi-epitope vaccine we combine the acceptable epitopes in a text document which is known as mapping of the vaccine construct. Epitopes that display non-allergenic, nontoxic, and immunogenic features are selected for the vaccine construct. The mapping of the vaccine construct tells us about the percentage of each epitope B-cell, CTL, HTL present in it. The final construct is then validated for antigenic, allergenic, and physicochemical properties.
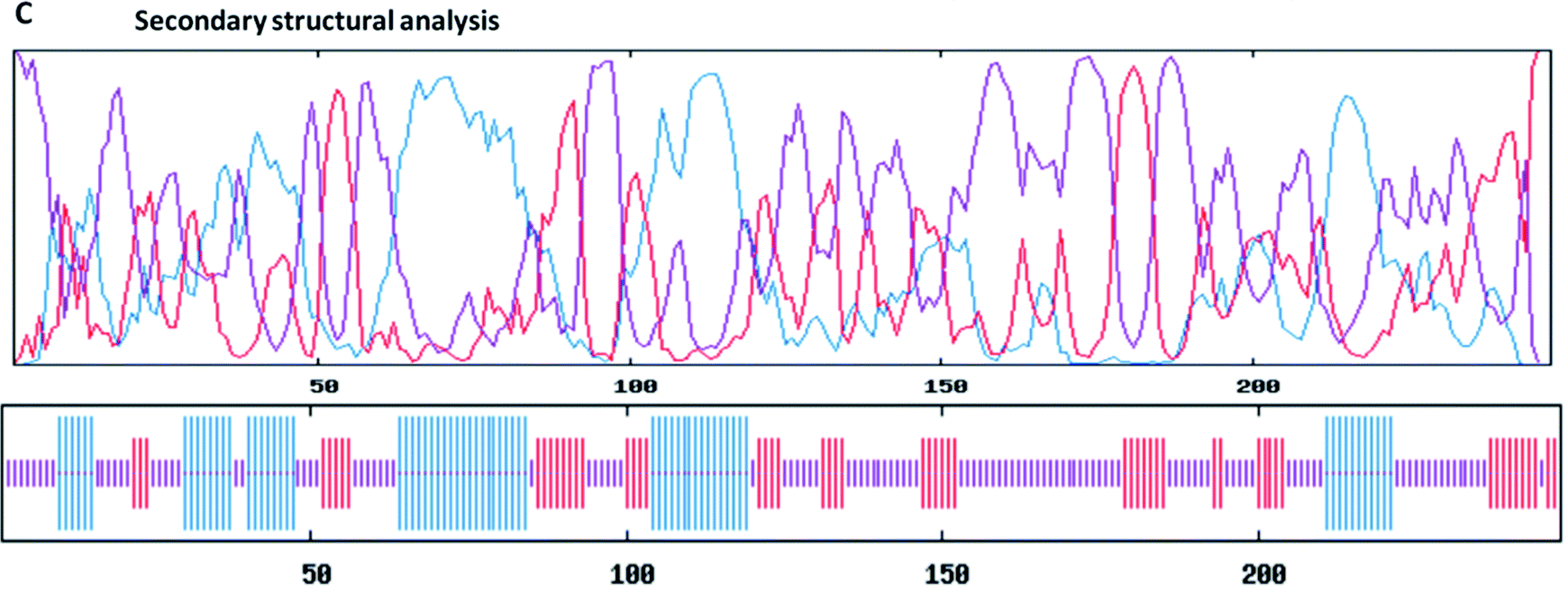
Perform Secondary and Tertiary Structure Prediction of Vaccines
To understand the 3D structure of vaccines, secondary structure should be predicted. The need for secondary and tertiary structure prediction is important in order to understand the proper functioning and allocation of vaccines in the cells. Vaccine production is an alternative approach to the traditional methods due to its safety, cost-effectiveness, stability, and specificity. Computational predictions of 2D and 3D structures of proteins using bioinformatics tools have made it easier to understand the working of proteins on hands by many researchers.
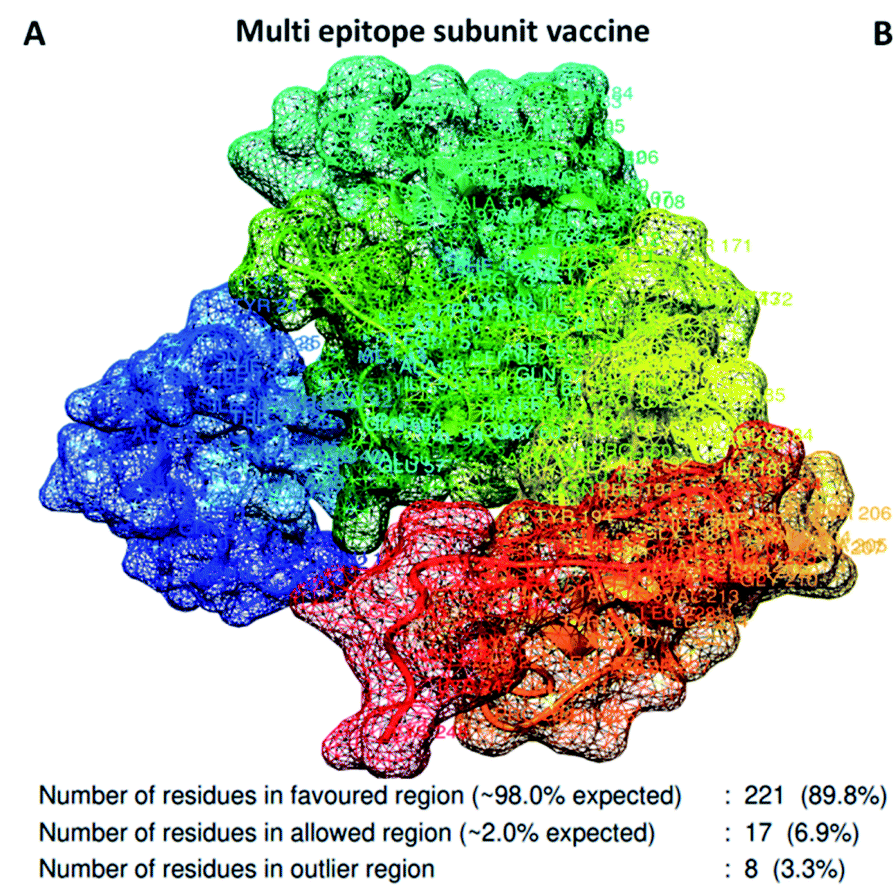
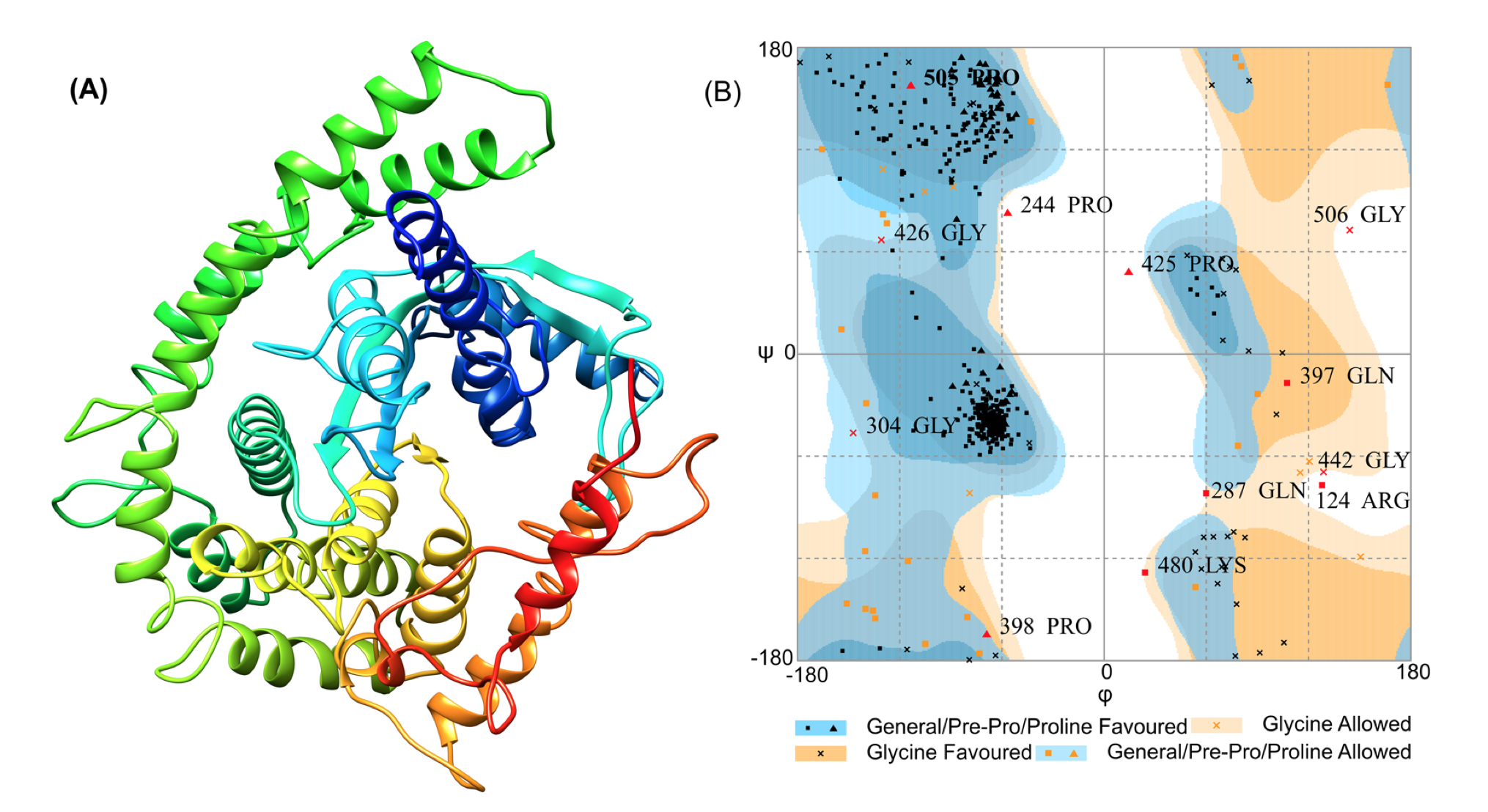
Tertiary Structure Refinement And Validation
The refinement of 3D models of vaccines has emerged as the last milestone of the structure prediction journey to reach parity with experimental accuracy. Refining 3D models often helps to bring them closer to native structures by modifying the secondary structure units and repacking sidechains. Computational predictions of 2D and 3D structures of proteins using bioinformatics tools have made it easier to validate refinements of proteins. Many reliable and extensively used computational methods are used to validate the quality of protein structures for instance PROSA Web, ERRAT, Verify 3D, PROCHECK etc.
Predict Discontinuous B-Cell Epitope Prediction.
Discontinuous epitopes refer to amino acid sequences which have a folded conformation. To predict the discontinuous B-cell epitope we will use IEDB Analysis Resource. This is done in ElliPro: Antibody Epitope Prediction tool available at IEDB. The refinement parameters for the validated and predicted model of vaccine construct are checked. The discontinuous B-cell epitopes are also cross checked with the actual structure to know whether the exact amino acids are present or not. This helps in deciding whether the vaccine is useful for further analysis or not.
Molecular Dynamics and Immune Simulation
To check the stability of the developed vaccine we perform molecular dynamics and immune simulations.
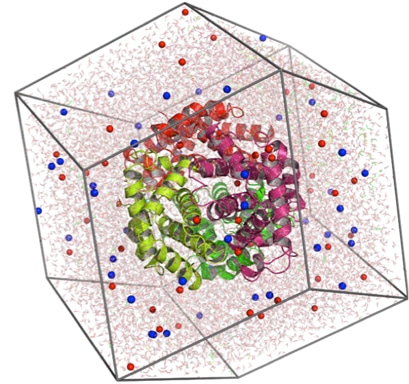
Perform Molecular Dynamic Simulation
Molecular dynamic simulation is the computational method that calculates the time dependent behavior of protein models for vaccine discovery. MS simulations have given detailed information on the fluctuations and conformational changes in the models. MD simulations are used with x-ray crystallography, cryo-electron microscopy, electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR) etc. Different tools that are used for molecular dynamic simulation are Abalone, CHARMM, GROMACS etc.
Perform Immune Simulation
Computational methods for the study of the immune system have grown to embed a great level of sophistication in the vaccine development process. The immune system is extremely complex with many interacting components determining overall outcomes. To check the immune simulation of a vaccine, C-Immsim 10.1 server can be used that combines systems biology techniques with the information provided by data-driven prediction methods.
Perform In-Silico Cloning
The identification of novel vaccines using database analysis is known as in silico cloning. In silico cloning may not only provide new information on genes and their biological function, it may also lead to identification of molecular targets for vaccine discovery activities. To conduct the in silico cloning of the vaccine, C-Immsim 10.1 server can be used.
Codon Optimization
Codon optimization is the process used to improve gene expression and increase the translational efficiency of a gene of interest by accommodating codon bias of the host organism so that stable codons are found. Molecular docking server HADDOCK (High Ambiguity Driven protein protein DOCKing) can be used to see the interaction of the designed vaccine and the receptor.
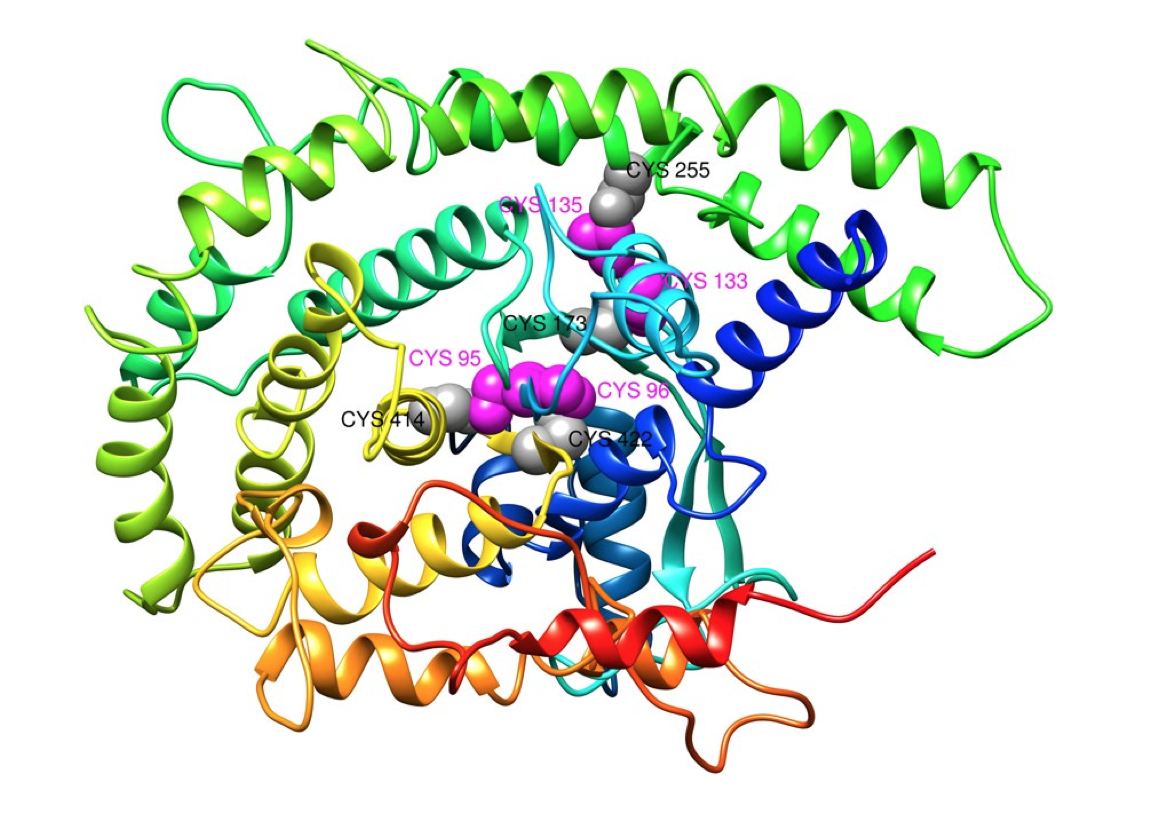
Disulfide Engineering
Disulfide Engineering is the modern approach to identify disulfide bonds in vaccine protein. These bonds give protein structure stability by confirming the precise geometry. These bonds prevent the protein structure from disruption that is strongly associated with loss of protein function and activity. Disulfide by Design 2 version 2.13 is a tool that can be used to engineer the Disulfide bonds in vaccine protein. It is a web-based, platform-independent application that significantly extends functionality, visualization, and analysis capabilities for disulfide engineering.
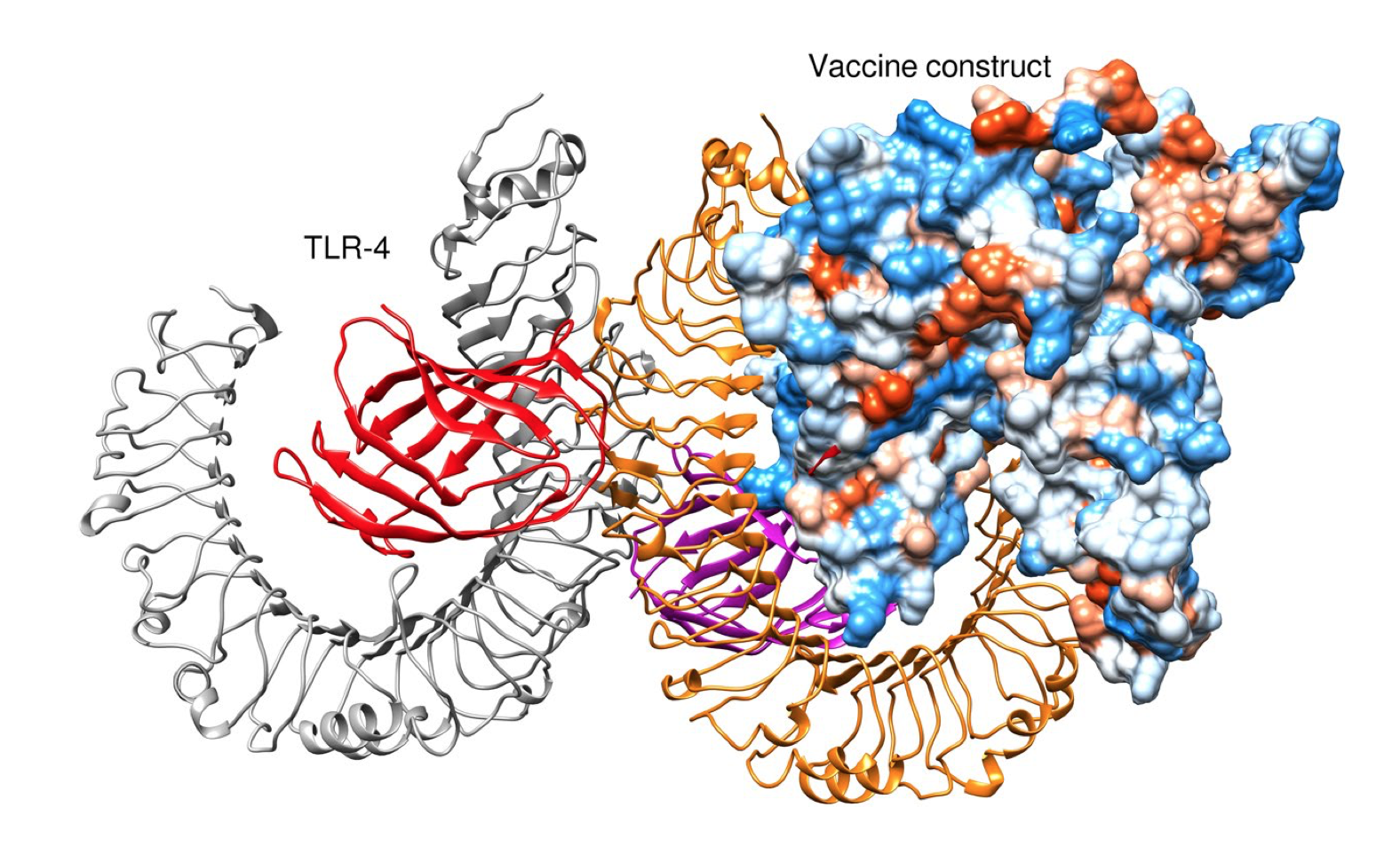
Molecular Docking between a Protein and Receptor
Molecular Docking is a bioinformatics modeling method which predicts the suitable orientation of a receptor to a second molecule that is protein when bound to each other to form a stable complex. Any molecular docking tool can be used to perform docking for instance MOE, AutoDock etc. HADDOCK version 2.4 is an institutional tool that distinguishes itself from ab-initio docking methods in the fact that it encodes information from identified or predicted protein interfaces in ambiguous interaction restraints (AIRs) to drive the docking process.
Get in touch with us and we will start your analysis right away!
