Introduction
The next-generation sequencing (NGS) has become one of the best approaches in the field of therapeutic diagnosis and treatment. With the help of NGS, complex pathophysiological diseases like cancer can have a potential drug for treatment. Globally, cancer is considered a complex disorder that is accompanied by lots of genetic mutations in germ cells as well as in somatic cells disturbing and deregulating the cell cycle.
In human beings 30,000 genes are present, among these genes, the potential pharmacological targets are found to be 6000 to 8000. Up till now, only 400 encoded proteins have been validated as an effective target for drug development. Compared to other illnesses, cancer has a varied amount of potential molecular targets that can be used for drug development. In traditional drug discovery, “one molecule – one target – one disease” is followed, as it does not take into account the interaction between proteins and the potential drugs. Due to this reason, cancer and many other diseases have been overlooked for drug treatment.
Besides this, poly-pharmacological properties of some drugs result in unexpected drug impacts that can produce undesirable side effects that cannot be controlled. This mostly happens in the case of cancer drugs. In many anticancer drugs, the target protein has not been identified. Also, in other instances, some of the potential targets like transcriptional factors, phosphatases, and RAS family members are considered undruggable as per pharmacological regulation. Thus, it has become a need in the development of anticancer drugs that all potential binding sites of a novel ligand must be characterized for drug repositioning. To help in this endeavor, high-throughput NGS with its bioinformatics tools can play an important role.
Next-Generation sequencing technologies are considered a high throughput sequencing and data analysis technique required for therapeutic cancer diagnosis and treatment. With the help of NGS technologies, tumor genomics can be assessed at the level of individual genes or transcripts through parallel sequencing and multiplexing of samples. The main challenge in cancer diagnosis and treatment is the heterogeneity of tumor cells which results in variability of treatment and also resistance development with time. With the help of NGS, we can identify the genetic alterations associated with tumor and resistance, which then helps in designing anticancer drugs for the specific patient with a precise dose for a defined time.
Identification of new cancer drug targets by NGS
Treatment of any type of cancer patient depends upon the decision which is based on the location of the tumor and the histological appearance. But it must be kept in mind that the response to the drug is mostly heterogeneous due to multiple alterations present in the same type of tumor. This molecular diversity is the main reason behind the ability of cancer to overcome various exogenous and endogenous control strategies. Thus, it is quite necessary to build an understanding of this cancer diversity and then manage it. NGS technologies have opened a gateway that cannot only help in understanding cancer heterogeneity but also helps in the development of anticancer drug according to individual requirements. These personalized treatments cannot only improve the control of tumors but also have fewer side effects.
Multiple NGS methodologies for cancer therapeutics
With the discovery of NGS technologies, early diagnosis of cancer is possible. This results in easier profiling of the cancer genome and gives information about the targeted therapy. To do genomic profiling of cancer patients, two pathological biopsy media techniques are employed i.e., paraffin embedding and formalin fixation. The profiling of cancer patients is influenced by steps involved in genomic data generated protocol that also includes pre-analytical techniques, library preparation, sequencing, and variant calling. The methods that are mostly employed in NGS are illustrated and explained below:
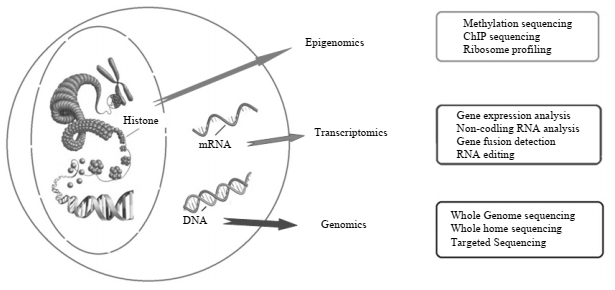
Figure 1: Multiple techniques of Next-generation sequencing technologies
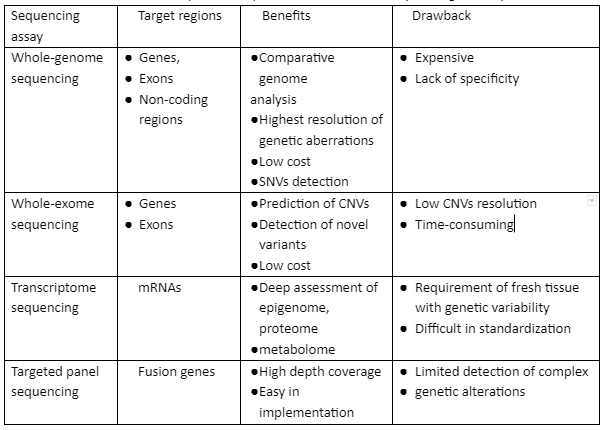
For understanding small mutations in the genome of cancer patients, techniques like whole-genome sequencing (WGS), transcriptome sequencing, whole-exome sequencing (WES), and targeted RNA panel sequencing are used. In WGS, copy number variants (CNV) are detected with very high resolution and regulatory sites like promoters and enhancers are determined. Also, it can find out about the role of intergenic regions in regulation. The researchers, with the help of WGS, can study cancer genomics for the identification and classification of some new mutations. On the other hand, WES has coverage of about 1-2% of the whole genome but it has 95% more coverage of the exon in comparison to WGS. The WES can identify germline and somatic alterations in cancer patients. The transcriptomic sequencing allows the genome profiling of cancer patients which helps in understanding the mRNA expression levels.
Transcriptome analysis cannot only determine gene expression but also can analyze DNA mutations, making it an effective technique. It also has very high significance for detection-based biomarker development through non-coding RNA (i.e., siRNA, miRNA, piRNA, and lncRNA). As a whole, transcriptome analysis can assess proteome, epigenome, and metabolome in a wider context.
Targeted panel sequencing is linked with precision-based cancer studies. In these techniques, alterations of genes have been determined in the 20-500 genes panel that is developed on amplicon-based or hybridization techniques. These techniques help detect single nucleotide variants (SNVs) and indels for cancer therapeutics. These techniques are quite useful in providing coverage with high exon and high depth which are two crucial aspects of variant calling. The coverage depth is defined as the repetition of some specific bases in sequencing and reference genome alignment. Besides this, exon coverage also shows almost one sequence read in percentage. In the targeted panel sequencing method, formalin-fixed embedded tissues have been used. Some of the features of WGS, WES, transcriptome sequencing, and targeted panel sequencing are given in table 1:
Table 1: Comparative aspects of common NGS sequencing techniques
Besides these techniques, other technologies employed in NGS are de-novo sequencing, epigenomics sequencing, and non-coding RNA sequencing. De-novo sequencing techniques generate a sequence of the complete genome by alignment of reads with themselves (in cases where a reference genome is unavailable). The epigenomic approach employed methylation sequencing which includes techniques like ChIP-sequencing and ribosome profiling. This sequencing methodology allows a deeper understanding of the regulation of gene expression profiles. It also allows us to identify methylation up to the level of single nucleotide and also DNA fragmentation up to 100-150 bp which is followed by standard libraries construction for carrying out NGS analysis. The Chromatin immune-precipitation (ChIP) sequencing allows the diagnosis of any disorder by the study of protein-RNA or protein DNA interaction. On the other hand, ribosome profiling is based on active mRNA fragments that are taken up by ribosomes in the translation process and they give us information about the cellular activity at a given time and also identify the active proteins that can modify different cellular processes. Lastly, non-coding RNA sequencing allows us to analyze the differential gene expression through repressors or silencers.
Basic Pipeline of NGS technologies
The type of NGS technology employed for analysis depends upon the therapeutic question and the sample type. The basic workflow of all the NGS technologies is the same but the treatment protocols and the diagnostic approach may differ. Some of the basic steps involved in NGS technologies are given as follows:
Step One: Sample/library preparation:
Genomic material is isolated from a diseased tissue is fragmented with the help of enzymes or mechanical means to make the required size fragment that can be used in the sequencer. Also, the amplification of the sample is preferred for almost 4-10 cycles by using PCR. The genomic information produced is in the form of ‘reads’ and it can be stored by using .FASTAQ or .FASTA or.SRA file formats.
Step Two: Quality check
In this step, the raw reads produced as the result of step one are first analyzed for quality control before applying any type of analysis because any bad quality reads will affect the downstream analysis. So, the bad quality reads are removed with the help of software like FastQC.
Step 3: Mapping/Alignment of reads
In this step, the alignment of sequence reads to the reference genome is done which help us to find overlapping zone and alterations. This is a sensitive step as the contigs are constructed and mapped to the reference genome. any problems in this step will lead to false-positive and false-negative results.
Step Four: Downstream analysis:
All the effort in NGS technologies is to understand the genome through downstream analysis. This analysis gives us the output depending on our therapeutic question. The analysis helps to determine novel gene expression patterns from the specific type of cell and specific type of therapeutic condition. Sequencing of DNA is mostly done to find homozygous and heterozygous SNPs and mutations, indels, CNVs, and structural variants. On the other hand, RNA-seq is used for the identification of differential gene expression, splicing variant regions, signaling pathways and networks, and also gene regulatory networks. Many tools have been developed for downstream analysis, these include cummeRbund75, SAMtools73, and GATK74.
In cancer studies, most tumors are the culmination of genetic mutations and alternations in specific cells. Also, these mutations and problems can be genetic or epigenetic. The variance of normal and tumor cells was described by using highly accurate and sensitive techniques like mpileup, SAMtools, Isaac variant caller, and GATK.
The workflow of NGS sequencing with linked software is illustrated in figure 2:
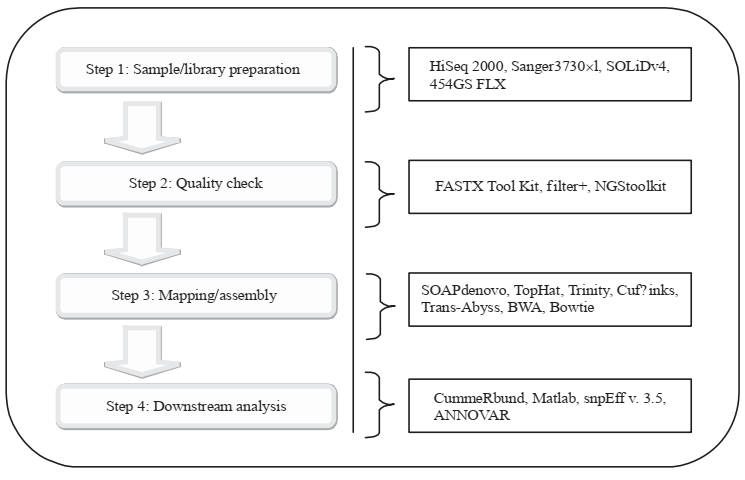
Figure 2: Workflow of next-generation sequencing technology with associated software
NGS and Target Capture Technology
As mentioned in the previous section, NGS has utilized many approaches for sequencing high throughput data and its analysis. The use of NGS in cancer required high performance and consistent diagnosis and treatment strategies. Through NGS, many biomarkers have been validated that were identified for cancer diagnosis as given in table 2:
Table 2: Globally identified cancer diagnostic biomarkers that are validated through NGS
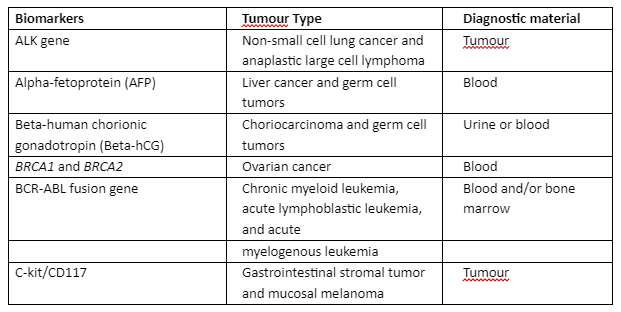
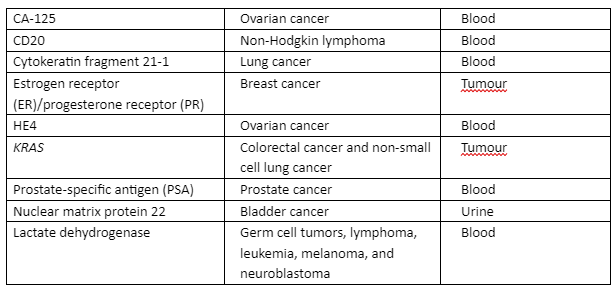
In traditional techniques of sequencing, a large amount of DNA or RNA sample was required for the analysis of the therapeutic target but with NGS, the analysis can be done with a small amount of genetic material and it can then be screened for several genes in a parallel way. Target Capture Technologies (TCT) is developed to make use of the known biomarkers and target a small potential set of therapeutic genes, TCT depends upon only two factors that are the type of sample and the quality and quantity of RNA or DNA. Some of the important TCT technologies are tabulated in table 3 with an underlying principle:
Table 3: Important TCT technologies with underlying methods and requirements

Shifting Paradigm: NGS Guided Personalized Oncogenic Treatment
The researchers have found several diagnostic methods for cancer through NGS identifications of novel mutations linked with it. Up till now, many oncogenic biomarkers have been discovered and also characterized in clinical practice by following the guideline of ʻThe American Society of Clinical Oncologyʼ (ASCO). These findings have enabled the researchers to move toward the field of personalized diagnosis and its relevant treatment. Thus, it is safe to say that NGS has revolutionized genome sequencing and also medical science by providing us with techniques for :
Rapid identifications of biomarkers
Whole genome sequencing or targeted gene sequencing for alternations and mutations
Identification of genetic interactions and associations
NGS has improved and developed over the years to become a more accurate and precise technique. The clinical ability of NGS is apparent through its capability to target germline mutations in patients who suffered from primary level of carcinoma. In another study, the gene signature associated with gastric cancer was identified and this can be used for personalized medicine.
NGS has made it possible to have a complete history of a patient’s cancer and then by analyzing it a particular treatment can be recommended.
Conclusion
As explained in the above sections, it has become evident that sequencing technologies play an important role in therapeutic diagnosis and treatment. NGS has provided us with a rapid, cost-effective, and real way of decoding the mysteries of diseases like cancer. NGS allows the analysis of inter-tumor and intra-tumor heterogeneity and then identifies biomarkers associated with each type of tumor, helping in diagnosis and drug development. But besides everything, still, a lot of work needs to be done to make NGS more accurate and make computational programs more user-friendly.
