We provide complete services for Molecular Docking Services
From data collection, preprocessing, quality control to transcription factor binding sites, histone modifications and much more.
Providing services to
worldwide clients
Datasets
analyzed
Qualified researchers and
Bioinformatics analysts
Satisfied orders processed
Our Services
- Protein-Small Molecule Docking Service
- Protein-Protein Docking Service
- Protein-DNA Docking Service
- Protein-Ligand Docking Service
- Protein-Nucleic Acid Docking Service
- Antibody-Antigen Docking Service
- Protein-Peptide Docking Service
- Protein–Carbohydrate Docking Service
- Protein–Lipid Docking Service
- Reverse Docking Service
- Flexible Peptide Docking Service
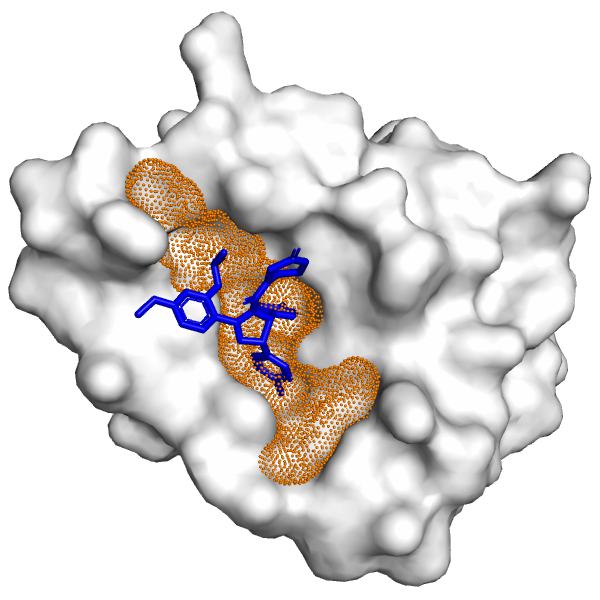
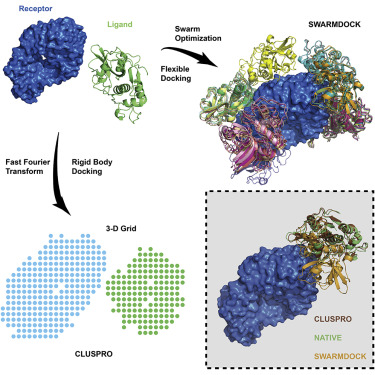
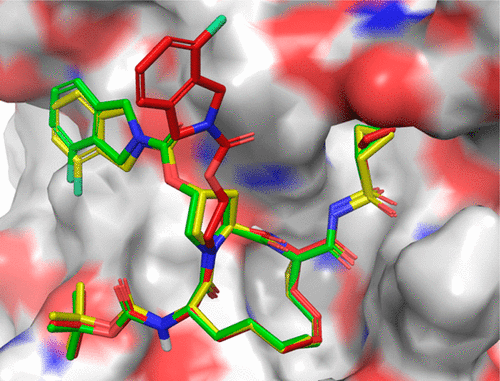
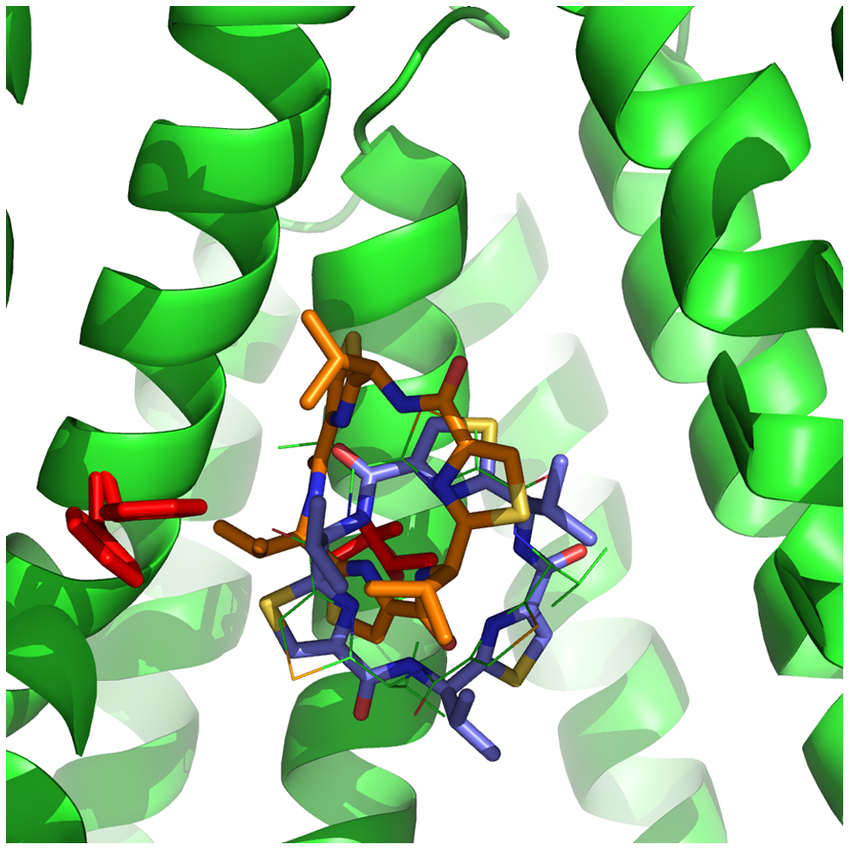
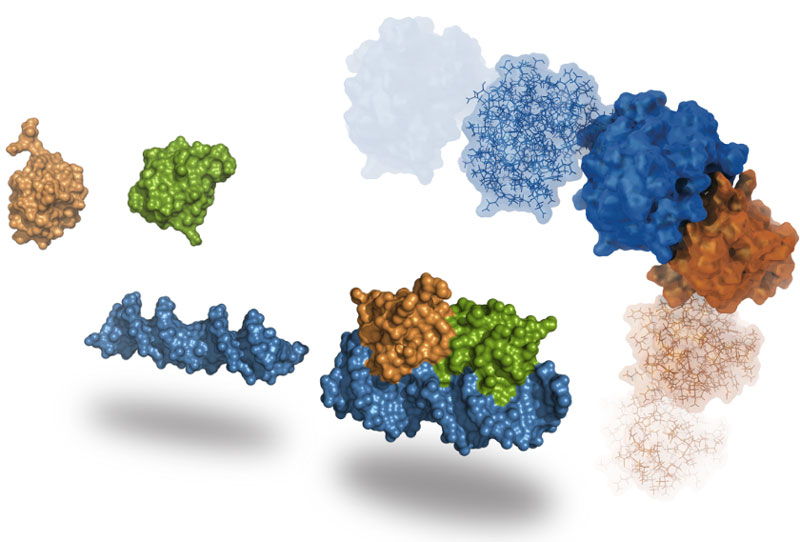
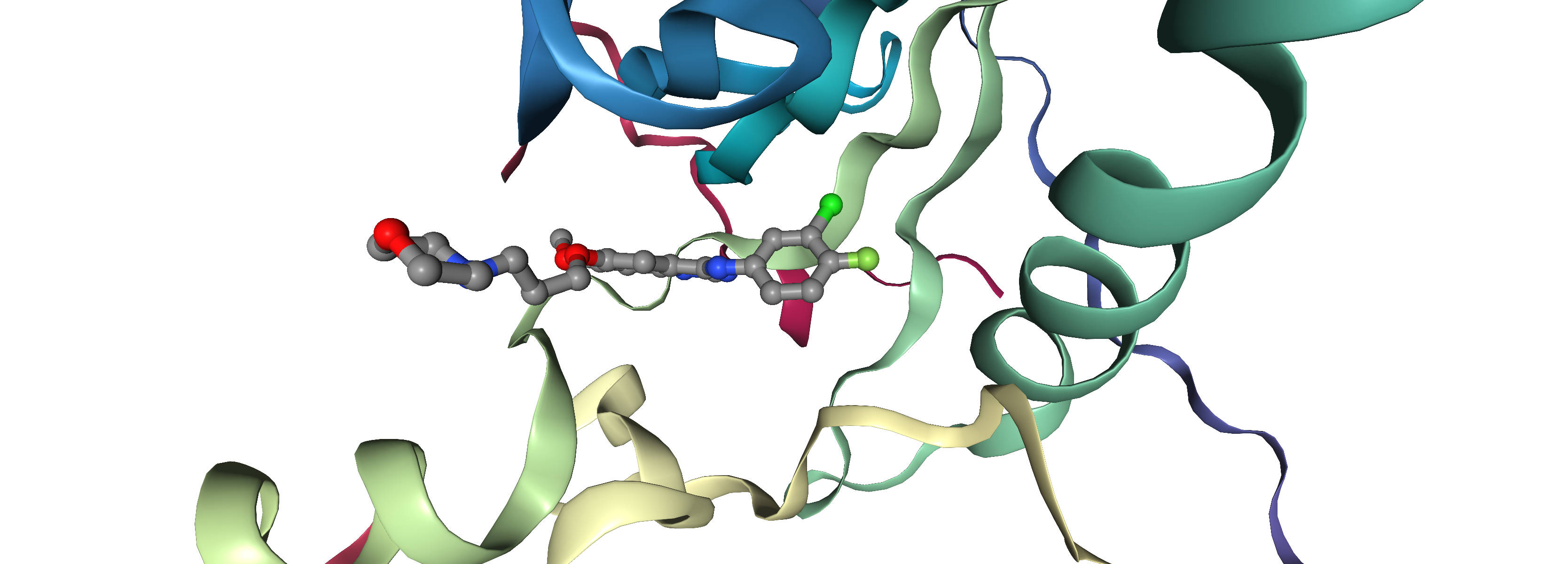
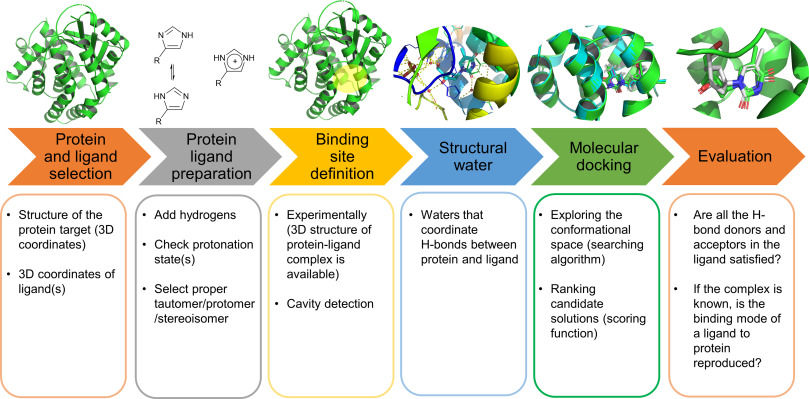
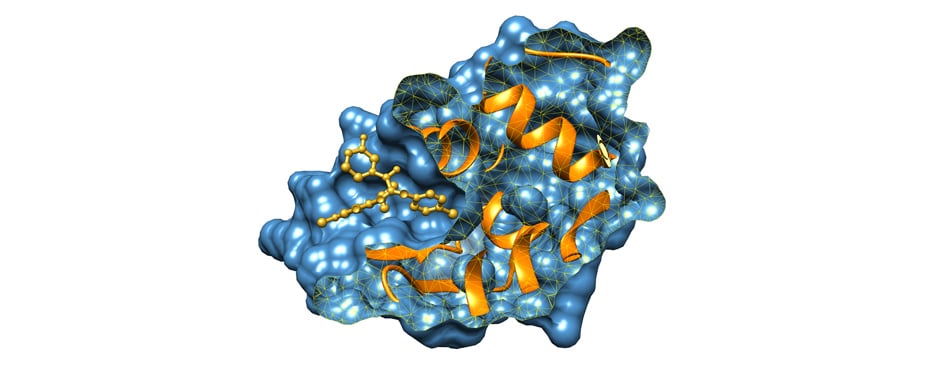
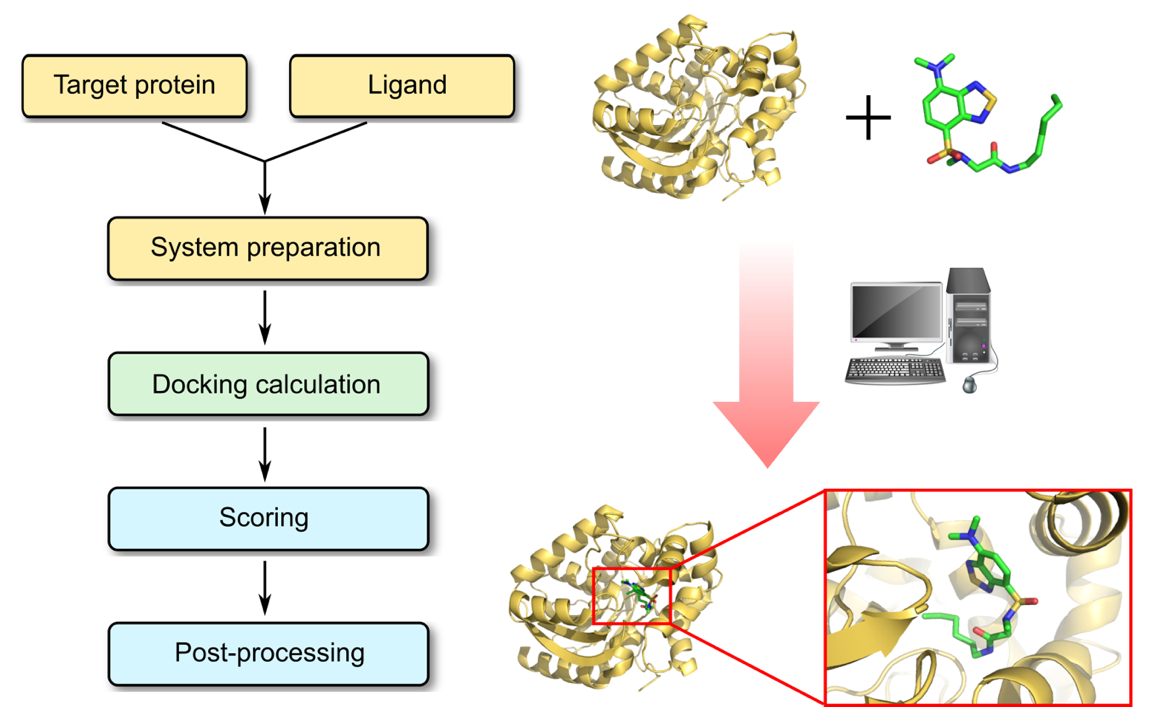
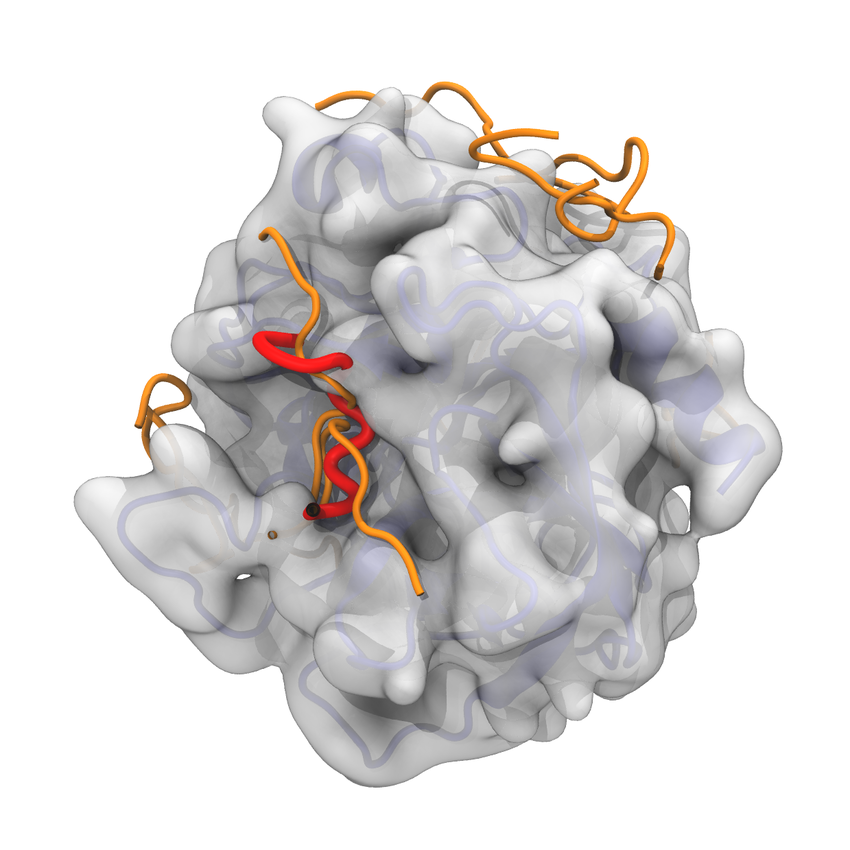
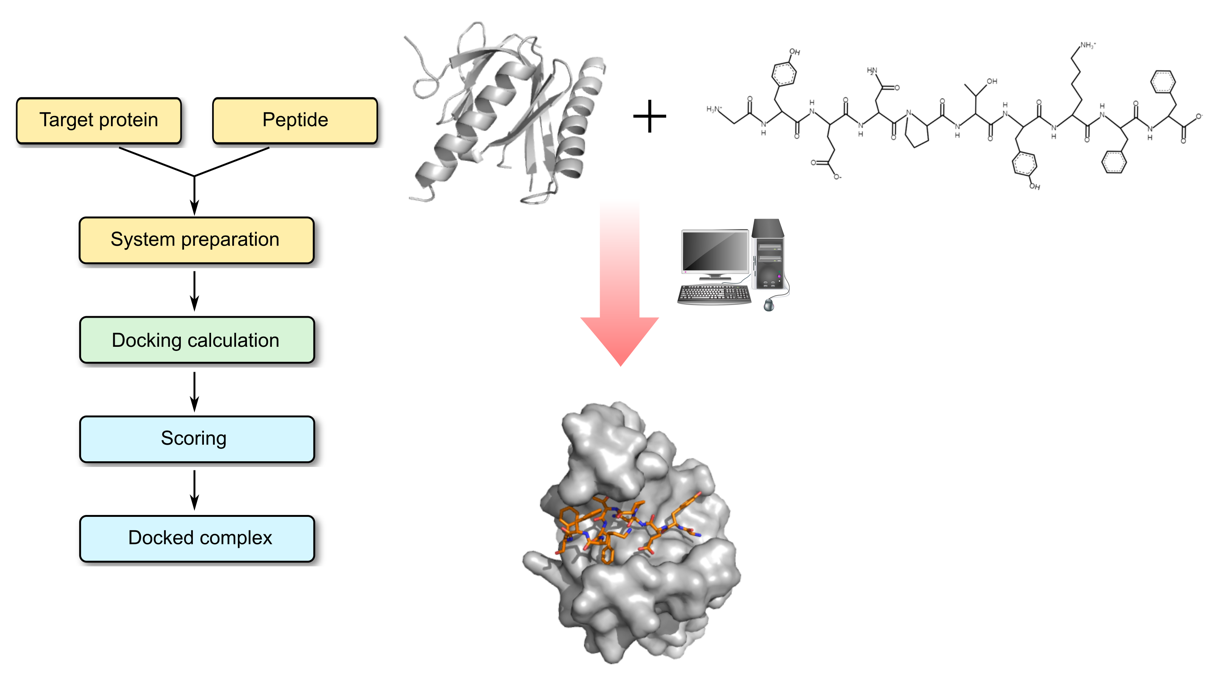
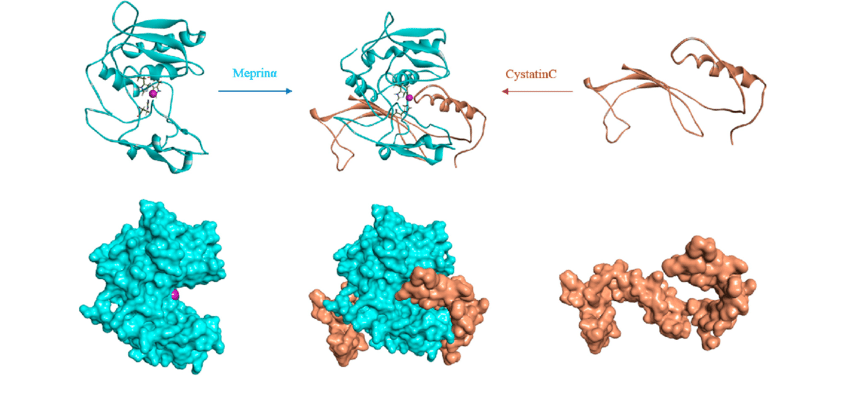
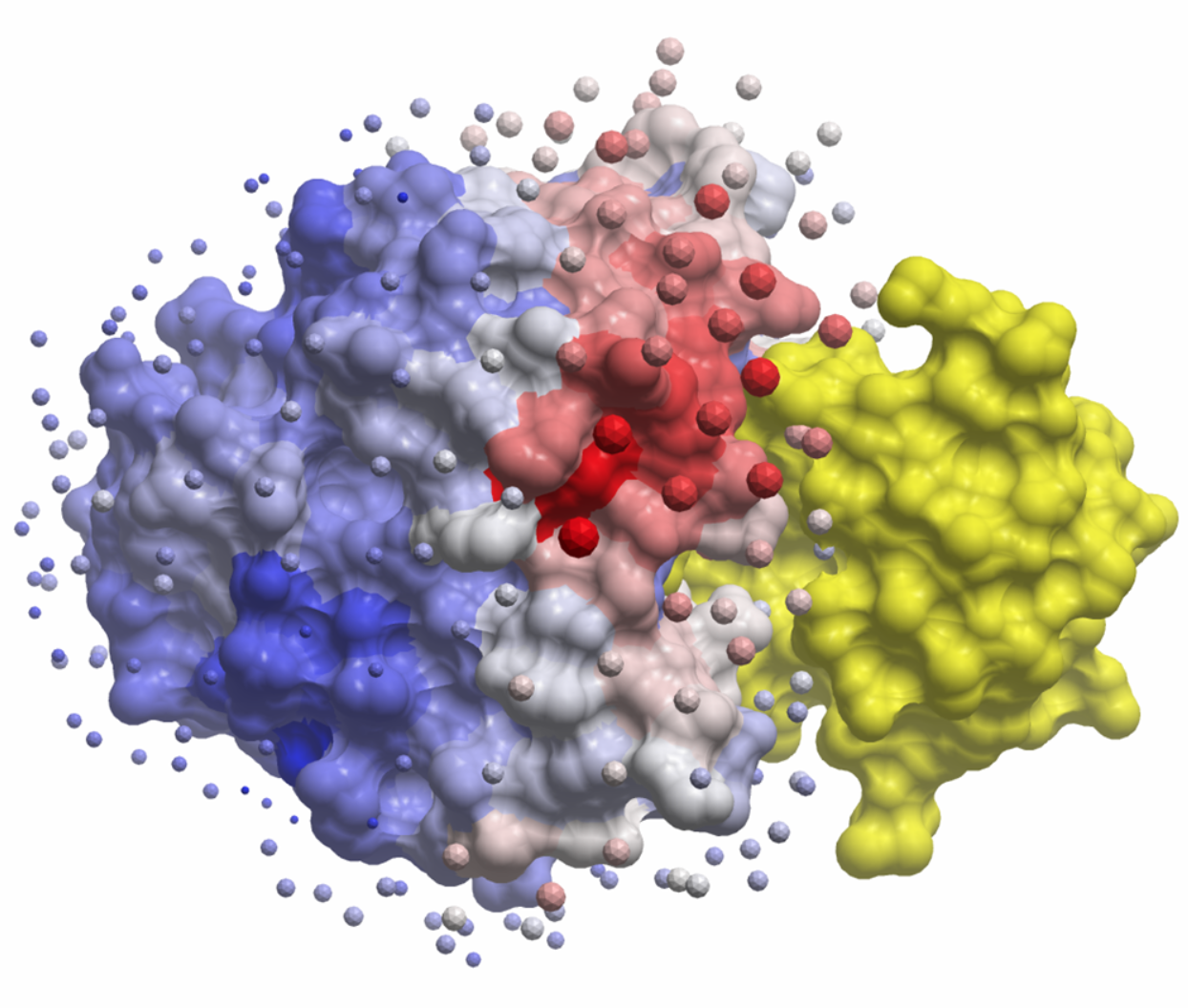
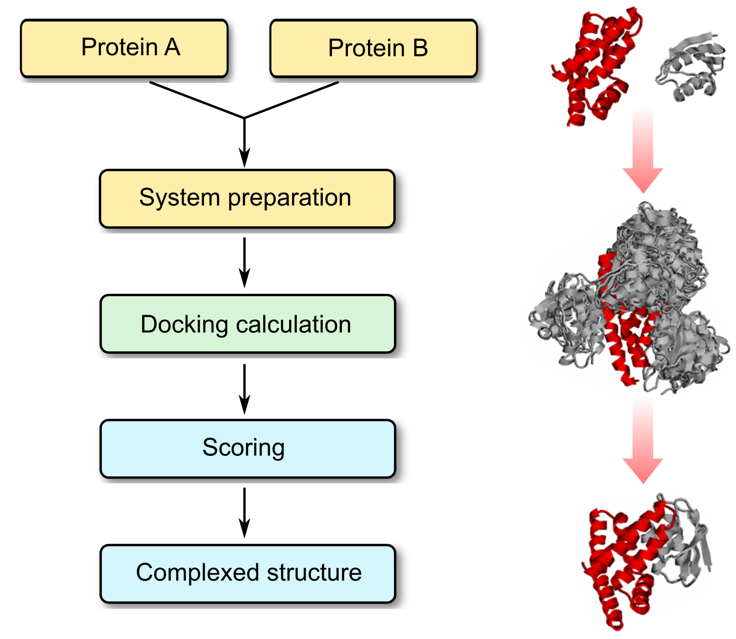
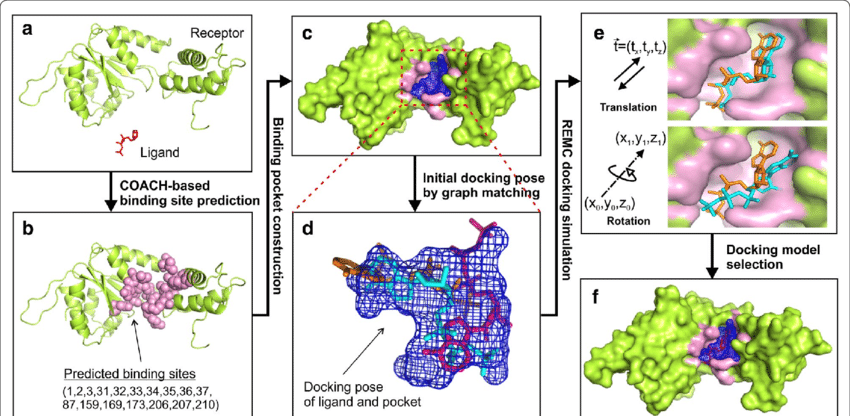
Molecular Docking is a bioinformatics modeling technique which involves the interaction of two or more molecules to give the stable adduct. Molecular docking is an important approach for designing new drugs and vaccines and other bioinformatics analysis as well. It predicts the three-dimensional structure of any complex depending upon binding properties of ligand and target. Molecular docking generates different possible adduct structures that are ranked and grouped together using a scoring function in the software.
Molecular interactions including protein-protein, enzyme-substrate, protein-nucleic acid, drug-protein, and drug-nucleic acid play important roles in many essential biological processes, such as signal transduction, transport, cell regulation, gene expression control, enzyme inhibition, antibody–antigen recognition and even the assembly of multi-domain proteins. These interactions very often lead to the formation of stable protein–protein or protein-ligand complexes that are essential to perform their biological functions.
Molecular docking classifies biomolecules into three categories: Proteins, Ligands and Peptides. The most important types of docking include protein-protein docking, protein-ligand docking and protein-peptide docking.
A Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. The protein-protein docking problem can be formulated as the prediction of the structure of the complex, given the structures of the individual proteins. Protein-protein docking aims to predict the structure of a protein-protein complex from its unbound components. An important step in protein-protein docking is the ranking of docked poses using a scoring function, for which many methods have been developed.
Protein-Ligand Docking
Ligands are the chemical compounds that interact with a biological target without disturbing its natural 3D conformation. Protein–ligand docking predicts the position and orientation of a ligand (a small molecule) when it is bound to a protein receptor or enzyme. Protein-ligand interactions are fundamental to many biological processes. The functions of a protein can be regulated by binding to ligands. A protein can have conformational changes under physiological conditions, which in turn are responsible for the conformational transitions among low- and high-affinity states for the ligands. Ligand binding can stabilize the protein in some specific states and thereby switch the protein among states of different functions.
Types of Protein-Ligand Docking
The mutual adaptation of a ligand with its receptor is crucial to understanding ligand binding and protein function. One of the major challenges in molecular docking is how to account for this adaptation in docking calculations.
The docking problem can be divided into following categories:
1. Rigid Body Docking: Rigid body docking ignores the flexibility of the molecules and treats them like rigid objects. It explores only six degrees of translational and rotational freedom, hence excluding any kind of flexibility.
2. Rigid Receptor – Flexible Ligand Docking: In this type of docking only the ligand is treated as flexible, the receptor is rigid. Thus it considers only the conformational space of the ligand.
3. Flexible Receptor – Flexible Ligand Docking: In this type of docking both protein and ligand are treated as flexible. Flexible receptor and flexible ligand interactions help in mimicking the binding process in real life.
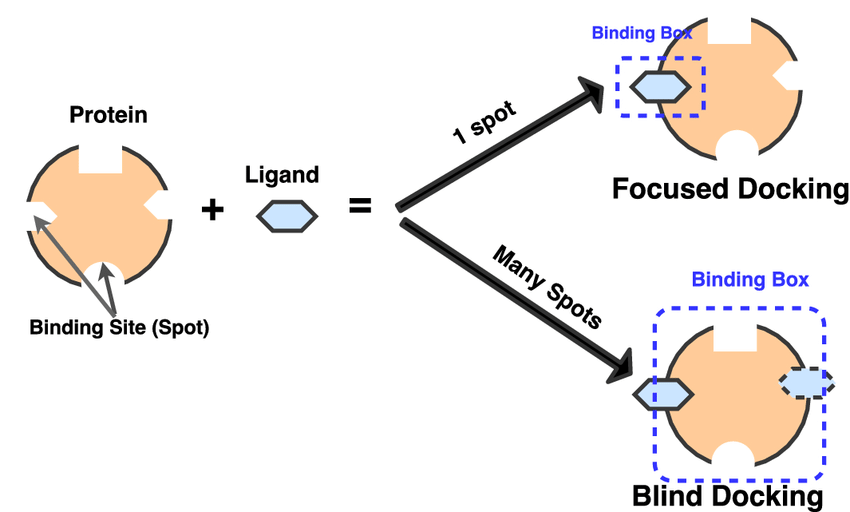
4. Blind Docking: Blind Docking refers to docking a ligand to the whole surface of a protein without any prior knowledge of the target pocket. Blind docking involves several trials/runs and several energy calculations before a favorable protein-ligand complex pose is found. However, the number of trials and energy evaluations necessary for a blind docking job is unknown.
Protein-Peptide Docking
A peptide is a short chain of amino acids. The amino acids in a peptide are connected to one another in a sequence by bonds called peptide bonds. Typically, peptides are distinguished from proteins by their shorter length, although the cut-off number of amino acids for defining a peptide and protein can be arbitrary. Peptides have recently attracted much attention as promising drug candidates. Rational design of peptide-derived therapeutics require structural characterization of the underlying protein-peptide interaction. Reliable computational tools are used for experimental characterization. In protein-peptide docking, first we predict the structure of the protein-peptide complex, starting from the protein structure and the peptide sequence, including variable degrees of information about the peptide binding site and/or conformation.
Tools and Servers Available for Molecular Docking
AutoDock Vina, SwissDock, rDock, LZerD, PatchDock, ZDock VirtualFlow, MOE (Molecular Operating Environment), HADDOCK and many others.
AutoDock Vina
AutoDock Vina is a successor of AutoDock, significantly improved in terms of accuracy and performance. AutoDock is a molecular modeling simulation software. It is especially effective for protein-ligand docking. AutoDock Vina was found to be a strong competitor against the other docking programs. It significantly improved the average accuracy of the binding mode predictions compared to AutoDock. AutoDock Vina is one of the most cited docking software applications in the research community.
SwissDock
SwissDock is a web service that predicts the molecular interactions that may occur between a target protein and a small molecule. It is used to propose a binding mode for a ligand, create figures for your articles, generate a complex to perform subsequent calculations and design inhibitors for the target of your choice.
MOE
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research.
PatchDock
The PatchDock server performs structure prediction of protein–protein and protein–small molecule complexes. PatchDock is a geometry-based molecular docking algorithm. It is aimed at finding docking transformations that yield good molecular shape complementarity.
rDock
rDock, previously known as RiboDock, is an open-source molecular docking software that can be used for docking small molecules against proteins and nucleic acids. It is primarily designed for high-throughput virtual screening and prediction of binding mode.
VirtualFlow
VirtualFlow is able to use a variety of the most powerful docking programs. VirtualFlow can access vast regions of the chemical space and identify molecules that bind with high affinity to target proteins.
HADDOCK
HADDOCK is a tool that distinguishes itself from ab-initio docking methods in the fact that it encodes information from identified or predicted protein interfaces in ambiguous interaction restraints (AIRs) to drive the docking process. It is an integrative platform for the modeling of biomolecular complexes. It supports a large variety of input data and can deal with multi-component assemblies of proteins, peptides, small molecules and nucleic acids.
Get in touch with us and we will start your analysis right away!
